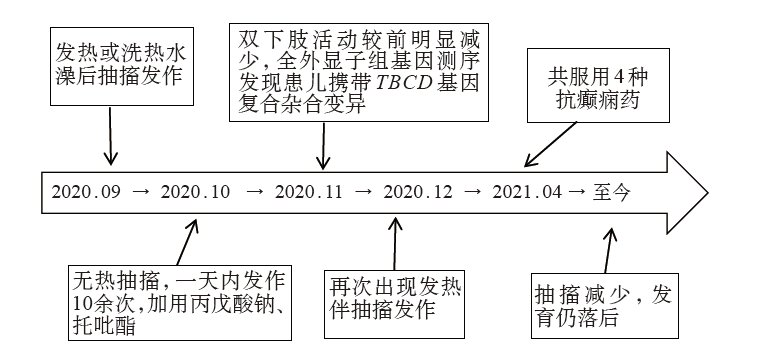
| [1] |
Miyake N, Fukai R, Ohba C, et al. Biallelic TBCD mutations cause early-onset neurodegenerative encephalopathy[J]. Am J Hum Genet, 2016, 99(4):950-961.
doi: 10.1016/j.ajhg.2016.08.005
|
| [2] |
Tian G, Lewis SA, Feierbach B, et al. Tubulin subunits exist in an activated conformational state generated and maintained by protein cofactors[J]. J Cell Biol, 1997, 138(4): 821-832.
pmid: 9265649
|
| [3] |
Stephen J, Nampoothiri S, Vinayan KP, et al. Cortical atrophy and hypofibrinogenemia due to FGG and TBCD mutations in a single family: a case report[J]. BMC Med Genet, 2018, 19(1): 80.
doi: 10.1186/s12881-018-0597-6
pmid: 29769041
|
| [4] |
Grønborg S, Risom L, Ek J, et al. A Faroese founder variant in TBCD causes early onset, progressive encephalopathy with a homogenous clinical course[J]. Eur J Hum Genet, 2018, 26(10): 1512-1520.
doi: 10.1038/s41431-018-0204-5
pmid: 29921875
|
| [5] |
Zhang Y, Zhang L, Zhou S. Developmental regression and epilepsy of infancy with migrating focal seizures caused by TBCD mutation: a case report and review of the literature[J]. Neuropediatrics, 2019, 51(1): 68-71.
doi: 10.1055/s-0039-1698423
|
| [6] |
Al-Bakheet A, Tohary M, Khan S, et al. Hematological findings associated with tubulin-folding cofactors D‐related encephalopathy: expanding the phenotype[J]. Clin Genet, 2021, 99(5): 724-731.
doi: 10.1111/cge.13932
pmid: 33506509
|
| [7] |
Ikeda T, Nakahara A, Nagano R, et al. TBCD may be a causal gene in progressive neurodegenerative encephalopathy with atypical infantile spinal muscular atrophy[J]. J Hum Genet, 2017, 62(4): 473.
doi: 10.1038/jhg.2016.149
|
| [8] |
Rudnik-Schöneborn S, Goebel HH, Schlote W, et al. Classical infantile spinal muscular atrophy with SMN deficiency causes sensory neuronopathy[J]. Neurology, 2003, 60(6): 983-987.
pmid: 12654964
|
| [9] |
Landrieu P, Baets J. Early onset (childhood) monogenic neuropathies[J]. Handbook Clin Neurol, 2013, 115:863-891.
|
| [10] |
Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies[J]. Brain, 2014, 137(Pt 11): 2879-2896.
doi: 10.1093/brain/awu169
|
| [11] |
Flex E, Niceta M, Cecchetti S, et al. Biallelic mutations in TBCD, encoding the tubulin folding cofactor D, perturb microtubule dynamics and cause early-onset encephalopathy[J]. Am J Hum Genet, 2016, 99(4):962-973.
doi: 10.1016/j.ajhg.2016.08.003
|
| [12] |
Bahi-Buisson N, Poirier K, Fourniol F, et al. The wide spectrum of tubulinopathies: what are the key features for the diagnosis?[J]. Brain, 2014, 137(Pt 6): 1676-1700.
doi: 10.1093/brain/awu082
pmid: 24860126
|
| [13] |
Okumura M, Sakuma C, Miura M, et al. Linking cell surface receptors to microtubules: tubulin folding cofactor D mediates dscam functions during neuronal morphogenesis[J]. J Neuroscience, 2015, 35(5): 1979-1990.
doi: 10.1523/JNEUROSCI.0973-14.2015
|
| [14] |
Cleveland DW, Yamanaka K, Bomont P. Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway[J]. Hum Mol Genet, 2009, 18(8): 1384-1394.
doi: 10.1093/hmg/ddp044
pmid: 19168853
|
| [15] |
宋彬彬, 陈峥, 贾炳泉, 等. 微管异常与肌萎缩侧索硬化症[J]. 神经损伤与功能重建, 2020, 15(4): 217-220.
|
 ), 郑帼
), 郑帼
 沪公网安备 31011002000392号
沪公网安备 31011002000392号