

Journal of Clinical Pediatrics ›› 2025, Vol. 43 ›› Issue (4): 278-285.doi: 10.12372/jcp.2025.24e0468
• Original Article • Previous Articles Next Articles
SUN Qiqing1, CHEN Mengmeng2, FU Dapeng1, HE Kun1, YAN Xiaochen1, HOU Weina1, WANG Fangjie1, XIE Zhenhua3( ), LI Dongxiao3
), LI Dongxiao3
Received:2024-05-10
Accepted:2024-11-27
Published:2025-04-15
Online:2025-03-31
Contact:
XIE Zhenhua
E-mail:xzhh0107@163.com
SUN Qiqing, CHEN Mengmeng, FU Dapeng, HE Kun, YAN Xiaochen, HOU Weina, WANG Fangjie, XIE Zhenhua, LI Dongxiao. Clinical and genetic analysis of 14 children with congenital long QT syndrome[J].Journal of Clinical Pediatrics, 2025, 43(4): 278-285.
Table 1
Clinical phenotype of 14 children with LQTS"
| 患儿 | 表型 | 性别 | 发病 年龄 /月 | 确诊 用时 /月 | 发病症状 | 诱因 | 心律失常 | 心电图表现 | 运动 平板试验 | Schwartz 评分 | 家族史 | 随访 结局 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 例1 | LQTS1 | 男 | 72 | 2 | 晕厥 | 运动 | 无 | QTc537ms | 未做 | 5 | 无 | 存活 |
| 例2 | LQTS1 | 女 | 114 | 24 | 晕厥 | 运动 | 无 | QTc564ms | 未做 | 5 | 母亲及弟弟均有晕厥史 | 存活 |
| 例3 | LQTS1 | 男 | 82 | 36 | 晕厥 | 运动 | 无 | QTc508ms | 运动后 QTc534ms | 5 | 无 | 存活 |
| 例4 | LQTS1 | 女 | 168 | 1 | 晕厥 | 运动 | 无 | QTc518ms | 未做 | 5 | 母亲QTc延长,无心脏事件 | 存活 |
| 例5 | LQTS1 | 女 | 83 | 3 | 晕厥 | 运动 | 无 | QTc475ms | 运动后 QTc537ms | 5 | 母亲QTc延长,无晕厥或心脏骤停 | 存活 |
| 例6 | LQTS1 | 女 | 159 | 0 | 心跳骤停 | 运动 | 室速、室颤 | QTc588ms | 未做 | 6 | 无 | 死亡 |
| 例7 | LQTS2 | 男 | 3 | 0 | 心跳骤停 | 哭闹 | 室速、室早 | QTc495ms | 未做 | 5 | 母亲及哥哥QTc延长,无心脏事件 | 存活 |
| 例8 | LQTS2 | 男 | 104 | 0 | 阿斯综合征、心跳骤停 | 感染 | 室速、室颤 | QTc564ms | 未做 | 5 | 父亲QTc延长,无心脏事件 | 存活 |
| 例9 | LQTS2 | 男 | 159 | 1 | 阿斯综合征 | 睡眠 | 无 | QTc481ms,T波有切迹 | 未做 | 4 | 母亲QTc延长,无心脏事件 | 存活 |
| 例10 | LQTS3 | 女 | 0.3 | 0 | 心跳骤停 | 睡眠、哭闹 | 室早室速及室颤 | QTc524ms | 未做 | 5 | 无 | 存活 |
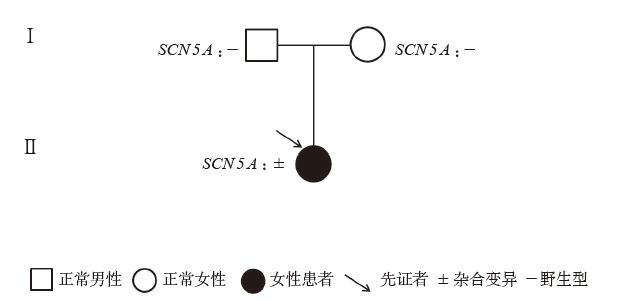
| 例11 | LQTS3 | 女 | 120 | 0 | 阿斯综合征、心跳骤停 | 睡眠、安静状态 | 室速、室颤 | QTc623ms | 未做 | 5 | 母亲 QTc延长,有晕厥史 | 存活 |
| 例12 | LQTS8 | 男 | 52 | 1 | 阿斯综合征 | 睡眠 | 无 | QTc553ms,2:1AVB,心率慢 | 未做 | 3.5 | 无 | 死亡 |
| 例13 | LQTS8 | 女 | 45 | 0 | 阿斯综合征、心跳骤停 | 哭闹 | 室速、室颤 | QTc596ms | 未做 | 5 | 无 | 死亡 |
| 例14 | LQTS14 | 女 | 15 | 54 | 阿斯综合征、晕厥 | 运动或情绪激动 | 无 | QTc496ms | 运动后 QTc509ms | 5 | 无 | 存活 |
Table 2
Genetic test results of 13 children with LQTS"
| 患儿 | 致病基因 | 转录本 | 转录本版本号 | 突变 位置 | 基因变异 | 突变类型 | 突变来源 | ACMG 评级及证据 |
|---|---|---|---|---|---|---|---|---|
| 例1 | KCNQ1 | NM_000218.3 | ENST 00000155840.12 | exon7 | c.1022C>T (p.Ala341Val) | 错义突变 | 父亲 | 致病(PS3+PM1+PM5+ PP3+PM2_Supporting)[ |
| 例2 | KCNQ1 | NM_000218.3 | ENST 00000155840.12 | exon7 | c.1022C>T (p.Ala341Val) | 错义突变 | 母亲 | 致病(PS3+PM1+PM5+ PP3+PM2_Supporting)[ |
| 例3 | KCNQ1 | NM_000218.3 | ENST 00000155840.12 | exon7 | c.940G>A (p.Gly314Ser) | 错义突变 | 新发突变 | 致病(PS2+PS4_Supporting+ PM1+PM5+PP3+PM2_ Supporting)[ |
| 例4 | KCNQ1 | NM_000218.3 | ENST 00000155840.12 | exon7 | c.944A>G (p.Tyr315Cys) | 错义突变 | 母亲 | 致病(PS3+PS4_Supporting+ PM1+PM5+PP3+PM2_ Supporting)[ |
| 例5 | KCNQ1 | NM_000218.3 | ENST 00000155840.12 | exon7 | c.1032G>A (p.Ala344 =) | 同义突变 | 母亲 | 可能致病(PS4_Supporting+ PM1+PP3+PM2_ Supporting)[ |
| 例7 | KCNH2 | NM_000238.3 | ENST 00000262186.5 | exon7 | c.1682C>T (p.Ala561Val) | 错义突变 | 母亲 | 致病(PS3+PS4_Moderate+ PM1+PM2 +PP2+PP3)[ |
| 例8 | KCNH2 | NM_000238.3 | ENST 00000262186.5 | exon7 | c.1881C>G (p.Phe627Leu) | 错义突变 | 父亲 | 可能致病(PS1+PM1+PP3+ PM2_Supporting)[ |
| 例9 | KCNH2 | NM_000238.3 | ENST 00000262186.5 | exon13 | c.3103del (p.Arg1035Glyfs*22) | 移码突变 | 母亲 | 可能致病(PVS1+PM2_ Supporting)[ |
| 例10 | SCN5A | NM_198056.2 | ENST 00000443581.1 | exon7 | c.796C>G (p.Leu266Val) | 错义突变 | 新发突变 | 可能致病(PS2+PP3+ PM2_Supporting) |
| 例11 | SCN5A | NM_198056.2 | ENST 00000443581.1 | exon28 | c.4859C>T (p.Thr1620Met) | 错义突变 | 母亲 | 可能致病(PS3+PS4_ Supporting+PP3+PM2_ Supporting)[ |
| KCNH2 | NM_000238.3 | ENST 00000262186.5 | exon10 | c.2414T>C (p.Phe805Ser) | 错义突变 | 母亲 | 可能致病(PM5+PM2_ supporting+PP3_moderate +PP4)[ | |
| 例12 | CACNA1C | NM_001167625.1 | ENST 00000399634.1 | exon8 | c.1204G>A (p.Gly402Ser) | 错义突变 | 新发突变 | 可能致病(PS2+PP3+PM2_ Supporting)[ |
| 例13 | CACNA1C | NM_000719.7 | ENST 00000399655.6 | exon8 | c.1216G>A (p.Gly406Arg) | 错义突变 | 新发突变 | 致病(PS2+PS4_Supporting+ PP3+PS1+PM2_ Supporting)[ |
| 例14 | CALM1 | NM_006888.4 | ENST 00000356978.4 | exon5 | c.395A>G (p.Asp132Gly) | 错义突变 | 新发突变 | 致病(PS2+PM1+PM5+ PP3+PM2_Supporting)[ |
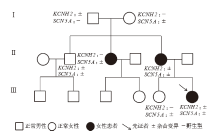
Figure 1
Genogram of case 11"
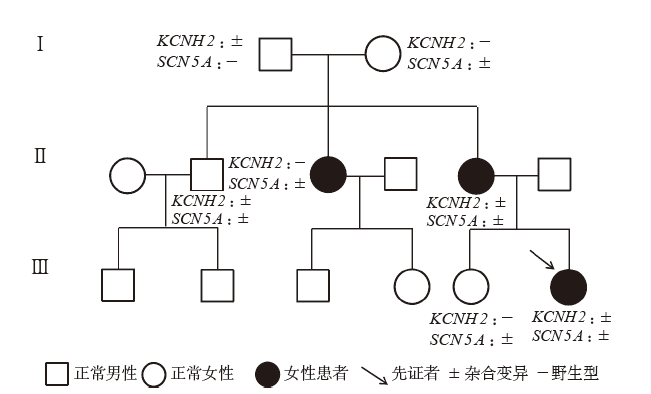
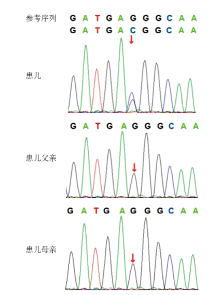
Figure 2
Genetic test results of case 10"
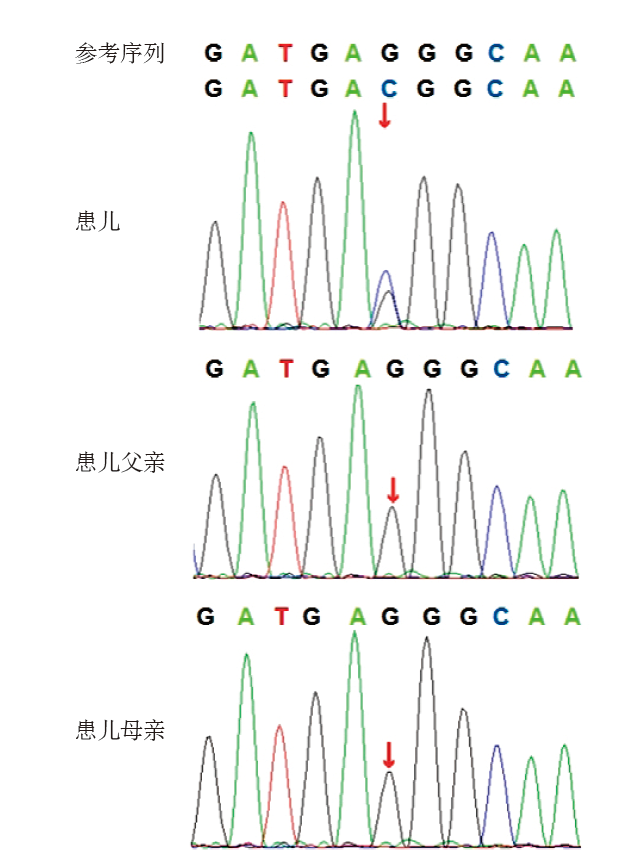
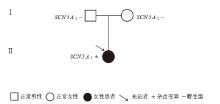
Figure 3
Genogram of case 10"
| [1] |
Steinberg C. Diagnosis and clinical management of long-QT syndrome[J]. Curr Opin Cardiol, 2018, 33(1): 31-41.
doi: 10.1097/HCO.0000000000000465 pmid: 28984650 |
| [2] | Neira V, Enriquez A, Simpson C, et al. Update on long QT syndrome[J]. J Cardiovasc Electrophysiol, 2019, 30(12): 3068-3078. |
| [3] |
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013[J]. Heart Rhythm, 2013, 10(12): 1932-1963.
doi: 10.1016/j.hrthm.2013.05.014 pmid: 24011539 |
| [4] | Westphal DS, Burkard T, Moscu-Gregor A, et al. Reclassification of genetic variants in children with long QT syndrome[J]. Mol Genet Genomic Med, 2020, 8(9): e1300. |
| [5] |
Hofman N, Jongbloed R, Postema PG, et al. Recurrent and founder mutations in the Netherlands: the long-QT syndrome[J]. Neth Heart J, 2011, 19(1): 10-16.
doi: 10.1007/s12471-010-0046-4 pmid: 21350584 |
| [6] |
Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test[J]. Heart Rhythm, 2009, 6(9): 1297-1303.
doi: 10.1016/j.hrthm.2009.05.021 pmid: 19716085 |
| [7] | Marschall C, Moscu-Gregor A, Klein HG. Variant panorama in 1,385 index patients and sensitivity of expanded next-generation sequencing panels in arrhythmogenic disorders[J]. Cardiovasc Diagn Ther, 2019, 9(Suppl 2): S292-S298. |
| [8] | Ng CA, Perry MD, Liang W, et al. High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants[J]. Heart Rhythm, 2020, 17(3): 492-500. |
| [9] |
Fazelifar AF, Pourirahim M, Masoumi T, et al. Identification of a novel pathogenic variant in KCNH2 in an Iranian family with long QT syndrome 2 by whole-exome sequencing[J]. J Arrhythm, 2023, 39(3): 430-453.
doi: 10.1002/joa3.12857 pmid: 37324772 |
| [10] |
Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing[J]. Heart Rhythm, 2010, 7(1): 33-46.
doi: 10.1016/j.hrthm.2009.09.069 pmid: 20129283 |
| [11] |
Fukuyama M, Ohno S, Ozawa J, et al. High prevalence of late-appearing T-wave in patients with long QT syndrome type 8[J]. Circ J, 2020, 84(4): 559-568.
doi: 10.1253/circj.CJ-19-1101 pmid: 32161207 |
| [12] | Sun Q, Xie Z, Wang F, et al. Case report of a child with long QT syndrome type 14 caused by CALM1 gene mutation and literature review[J]. Mol Genet Genomic Med, 2024, 12(1): e2287. |
| [13] |
Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death[J]. Eur Heart J, 2022, 43(40): 3997-4126.
doi: 10.1093/eurheartj/ehac262 pmid: 36017572 |
| [14] |
Barsheshet A, Dotsenko O, Goldenberg I. Congenital long QT syndromes: prevalence, pathophysiology and management[J]. Paediatr Drugs, 2014, 16 (6): 447-456.
doi: 10.1007/s40272-014-0090-4 pmid: 25288402 |
| [15] | Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy[J]. Heart Rhythm, 2019, 16(11):e301-e372. |
| [16] |
Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes[J]. Transl Res, 2013, 161(1): 1-14.
doi: 10.1016/j.trsl.2012.08.005 pmid: 22995932 |
| [17] | Huang H, Kuenze G, Smith JA, et al. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations[J]. Sci Adv, 2018, 4(3): eaar2631. |
| [18] |
Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism--a sex-specific modifier in type 1 long QT syndrome?[J]. BMC Med Genet, 2011, 12: 11.
doi: 10.1186/1471-2350-12-11 pmid: 21244686 |
| [19] | Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome[J]. N Engl J Med, 2003, 348(19): 1866-1874. |
| [20] |
Barsheshet A, Goldenberg I, J OU, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to β-blocker therapy in type 1 long-QT syndrome[J]. Circulation, 2012, 125(16): 1988-1996.
doi: 10.1161/CIRCULATIONAHA.111.048041 pmid: 22456477 |
| [21] | Copier JS, Bootsma M, Ng CA, et al. Reclassification of a likely pathogenic Dutch founder variant in KCNH2; implications of reduced penetrance[J]. Hum Mol Genet, 2023, 32(7): 1072-1082. |
| [22] |
Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing[J]. Heart Rhythm, 2005, 2(5): 507-517.
doi: 10.1016/j.hrthm.2005.01.020 pmid: 15840476 |
| [23] | 中华医学会心血管病学分会精准心血管病学学组, 中国医疗保健国际交流促进会精准心血管病分会, 中华心血管病杂志编辑委员会. 单基因遗传性心血管疾病基因诊断指南[J]. 中华心血管病杂志, 2019, 47(3): 175-196. |
| Section of Precision Cardiovascular Medicine of Chinese Society of Cardiology; Precision Cardiovascular Medicine Branch of China International Exchange; Promotive Association for Medical; Health Care Editorial Board of Chinese Journal of Cardiology. Guideline for the genetic diagnosis of monogenic cardiovascular diseases[J]. Zhonghua Xinxueguanbing Zazhi, 2019, 47(3): 175-196. | |
| [24] | 中国生物医学工程学会心律分会. 中国儿童心血管植入性电子器械专家共识[J]. 中国心脏起搏与心电生理杂志, 2023, 37(1): 1-11. |
| [25] |
Rohatgi RK, Sugrue A, Bos JM, et al. Contemporary outcomes in patients with long QT syndrome[J]. J Am Coll Cardiol, 2017, 70(4): 453-462.
doi: S0735-1097(17)37519-8 pmid: 28728690 |
| [1] | YAN Ge, HOU Cuilan, XIAO Tingting. Progress in mechanisms, diagnosis and therapeutic management of catecholaminergic polymorphic ventricular tachycardia [J]. Journal of Clinical Pediatrics, 2025, 43(3): 220-225. |
| [2] | LIU Qingyu, WANG Liwei, LIN Yilin, XIAO Rui, ZHOU Hui, ZHANG Xiaoqian, FU Mengran, MI Hongying. Genetic variation analysis of neonatal hyperbilirubinemia: a single-center retrospective study [J]. Journal of Clinical Pediatrics, 2024, 42(9): 782-786. |
| [3] | QI Qingwei. Promoting the application of new genetic testing technologies in the screening of fetal diseases with a scientific attitude [J]. Journal of Clinical Pediatrics, 2024, 42(5): 384-389. |
| [4] | WANG Ruifang, LIANG Lili, ZHANG Kaichuang, YANG Yi, SUN Yuning, SUN Manqing, XIAO Bing, HAN Lianshu, ZHANG Huiwen, GU Xuefan, YU Yongguo, QIU Wenjuan. Clinical characteristics and genetic variation analysis of Shwachman-Diamond syndrome in seven children [J]. Journal of Clinical Pediatrics, 2024, 42(3): 230-237. |
| [5] | YANG Zhibo, LIU Li. Application of gene sequencing technology in precise diagnosis and mechanism research of monogenic lupus [J]. Journal of Clinical Pediatrics, 2023, 41(9): 715-720. |
| [6] | TANG Yanan, YE Xiantao, GU Xuefan, YU Yongguo, XIAO Bing, SUN Yu. Clinical characteristics and genetic analysis in Chinese patients with Menke-Hennekam syndrome [J]. Journal of Clinical Pediatrics, 2023, 41(8): 613-617. |
| [7] | WANG Hongxia, PAN Xiang, LU Jun. Report a case of α-ketoadipic aciduria caused by compound heterozygous variant of DHTKD1 gene [J]. Journal of Clinical Pediatrics, 2023, 41(8): 624-628. |
| [8] | XIANG Chao, ZHANG Rong, KANG Lan, LEI Xiaoping, LIU Xingqing, DONG Wenbin. Association of coefficient of glycemic variation and SNAPPE-Ⅱ with prognosis in critically ill neonates [J]. Journal of Clinical Pediatrics, 2023, 41(6): 430-435. |
| [9] | WAN Ruiping, HUANG Xiaofei, YE Xingguang, WU Yanling, DAI Jiemin, LIU Zhigang. Clinical characteristics and genetic analysis of SETBP1 haploinsufficiency disorder [J]. Journal of Clinical Pediatrics, 2023, 41(6): 450-454. |
| [10] | ZHONG Yaoyao, ZHANG Liqin, DU Wei, LU Weibing, LIU Tingting. Gene mutation analysis and long-term follow-up of 6-pyruvoyltetrahydropterin synthase deficiency in Qingdao [J]. Journal of Clinical Pediatrics, 2023, 41(2): 103-107. |
| [11] | HU Haili, LI Weidong, WANG Yan, SONG Wangsheng, MA Qingqing. Neonatal screening and gene variation analysis of primary carnitine deficiency in Hefei City [J]. Journal of Clinical Pediatrics, 2023, 41(10): 680-684. |
| [12] | ZHANG Wanqiao, YAN Lei, ZHU Lina, MA Xiuwei. Metabolic and genetic analysis of abnormal 3-hydroxyisovalerylcarnitine in children from northern China [J]. Journal of Clinical Pediatrics, 2023, 41(10): 692-696. |
| [13] | ZHAO Jinhua, TANG Jihong, HUANG Jing, XIAO Xiao, ZHANG Bingbing, XING Yujiao, SHI Xiaoyan. Progressive myoclonic epilepsy caused by SEMA6B gene variations: a case report [J]. Journal of Clinical Pediatrics, 2022, 40(9): 705-709. |
| [14] | MEI Daoqi, MEI Shiyue, WANG Xiaona, WANG Yuan, CHEN Guohong, YANG Zhixiao, CHEN Xiaoyi, ZHANG Yaodong, YANG Xiuan. Early-onset epileptic encephalopathy type 27 caused by missense variation of GRIN2B gene: a case report [J]. Journal of Clinical Pediatrics, 2022, 40(4): 300-305. |
| [15] | FANG Hongjun, YANG Sai, KUANG Xiaojun, JIANG Zhi, ZHOU Zhen, WANG Lijuan, WU Liwen, YANG Liming, LIU Shulei, LIAO Hongmei. Analysis of clinical phenotype and genotype in 171 children with Duchenne muscular dystrophy [J]. Journal of Clinical Pediatrics, 2022, 40(3): 189-195. |
|
||