

Journal of Clinical Pediatrics ›› 2022, Vol. 40 ›› Issue (3): 189-195.doi: 10.12372/jcp.2022.21e0624
• Nervous System Disease • Previous Articles Next Articles
FANG Hongjun, YANG Sai, KUANG Xiaojun, JIANG Zhi, ZHOU Zhen, WANG Lijuan, WU Liwen, YANG Liming, LIU Shulei, LIAO Hongmei( )
)
Received:2021-04-26
Published:2022-03-15
Online:2022-03-09
Contact:
LIAO Hongmei
E-mail:hnliaohm006@sina.cn
FANG Hongjun, YANG Sai, KUANG Xiaojun, JIANG Zhi, ZHOU Zhen, WANG Lijuan, WU Liwen, YANG Liming, LIU Shulei, LIAO Hongmei. Analysis of clinical phenotype and genotype in 171 children with Duchenne muscular dystrophy[J].Journal of Clinical Pediatrics, 2022, 40(3): 189-195.
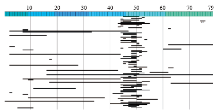
"
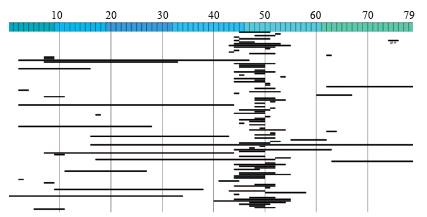
"
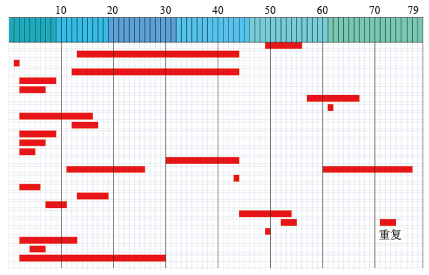
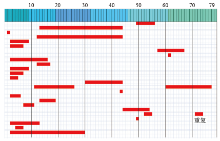
"
| 编号 | 变异信息 | 蛋白质改变 | 外显子 | ACMG评级 | 来源 | 数据库收录 | 变异 |
|---|---|---|---|---|---|---|---|
| 1 | c.3747G>A | p.W1249X | exon27 | 致病性 | 新发 | 已报道 | 错义变异 |
| 2 | c.7672C>T | p.Q2558* | exon53 | 致病性 | 新发 | 已报道 | 无义变异 |
| 3 | c.3625delC | p.Q1209Kfs*6 | exon27 | 致病性 | 新发 | 未报道 | 移码变异 |
| 4 | c.6117G>T | p.K2039N | exon42 | 临床意义未明 | 母源 | 未报道 | 错义变异 |
| 5 | c.9337C>T | p.R3113* | exon64 | 致病性 | 母源 | 已报道 | 无义变异 |
| 6 | c.1663C>T | p.Q555* | exon14 | 致病性 | 母源 | 已报道 | 无义变异 |
| 7 | c.9084+1G>A | splice-5 | intron60 | 致病性 | 母源 | 未报道 | 剪接变异 |
| 8 | c.1332-8A>G | splice | intron11 | 临床意义未明 | 母源 | 未报道 | 剪接变异 |
| 9 | c.1990C>T | p.Q664* | exon16 | 致病性 | 母源 | 未报道 | 无义变异 |
| 10 | c.4909_4910insGG | p.A1637Gfs*3 | exon35 | 致病性 | 新发 | 未报道 | 移码变异 |
| 11 | c.9204_9207delCAAA | p.N3068Kfs*20 | exon62 | 致病性 | 母源 | 已报道 | 移码变异 |
| 12 | c.2996T>C | p.L999P | exon23 | 临床意义未明 | 母源 | 未报道 | 错义变异 |
| 13 | c.3604-6T>A | splice | intron26 | 临床意义未明 | 母源 | 未报道 | 剪接变异 |
| 14 | c.10171C>T | p.R3391* | exon70 | 致病性 | 母源 | 已报道 | 无义变异 |
| 15 | c.956C>G | p.S319* | exon9 | 致病性 | 母源 | 已报道 | 无义变异 |
| 16 | c.8038C>T | p.R2680* | exon55 | 致病性 | 母源 | 未报道 | 无义变异 |
| 17 | c.4906G>T | p.E1636* | exon35 | 致病性 | 母源 | 未报道 | 无义变异 |
| 18 | c.4786_4787delAA | p.K1596Efs*5 | exon34 | 致病性 | 新发 | 已报道 | 移码变异 |
| 19 | c.7378G>T | p.(Glu2460*) | exon51 | 致病性 | 新发 | 未报道 | 无义变异 |
| 20 | c.1149G>T | p.(Glu383Asp) | exon10 | 临床意义未明 | 母源 | 已报道 | 错义变异 |
| 21 | c.4813dupA | p.(Ser1605fs) | exon34 | 致病性 | — | 未报道 | 移码变异 |
| 22 | c.1990C>T | p.(Gln664*) | exon16 | 致病性 | — | 已报道 | 无义变异 |
| 23 | c.9649+3A>C | splice | intron66 | 临床意义未明 | — | 未报道 | 剪接变异 |
| 24 | c.2758 C>T | p.(Gln920*) | exon21 | 致病性 | — | 已报道 | 无义变异 |
| 25 | c.187-2A>T | splice | intron3 | 致病性 | — | 未报道 | 剪接变异 |
| 26 | c.10603-10622delinsTGATG | p.(Gly3535*) | exon75 | 致病性 | — | 未报道 | 无义变异 |
| 27 | c.6614+1G>C | splice | intron45 | 致病性 | 新发 | 未报道 | 剪接变异 |
| 28 | c.9100C>T | p.(Arg3034*) | exon61 | 致病性 | — | 已报道 | 无义变异 |
| 29 | c.9337C>T | p.(Arg3113*) | exon64 | 致病性 | 新发 | 已报道 | 无义变异 |
| 30 | c.6677G>A | p.(Trp2226*) | exon46 | 致病性 | 母源 | 未报道 | 无义变异 |
| 31 | c.8937+2T>C | splice | intron59 | 致病性 | — | 已报道 | 剪接变异 |
| 32 | c.9258delC | p.(Met3088*) | exon63 | 致病性 | 母源 | 未报道 | 无义变异 |
| 33 | c.1201C>T | p.(Gln401*) | exon11 | 致病性 | 新发 | 已报道 | 无义变异 |
| 34 | c.1019delA | p.(Asn340fs) | exon10 | 致病性 | — | 未报道 | 移码变异 |
"
| 基因变 异类型 | 例数 | 男性[n(%)] | 年龄/岁 | LDH/IU·L-1 | CK/U·L-1 | CK-MB/U·L-1 | ALT/IU·L-1 | AST/IU·L-1 |
|---|---|---|---|---|---|---|---|---|
| 缺失 | 111 | 107(96.4) | 4.4 (2.0~7.0) | 1 016.0 (801.0~1 246.9) | 4 641.0 (1 308.0~9 546.4) | 178.4 (105.3~232.5) | 308.7 (229.3~413.8) | 198.8 (151.1~288.1) |
| 重复 | 26 | 24(92.3) | 4.7 (3.0~7.3) | 984.0 (803.0~1 152.5) | 3 501.0 (2 013.9~7 824.3) | 165.7 (86.4~259.9) | 261.8 (136.7~414.1) | 188.4 (123.3~268.0) |
| 错义变异 | 4 | 0(0.0) | 3.3 (1.3~3.6) | 617.0 (431.0~1 201.0) | 2 842.3 (2 632.3~5 882.4) | 189.4 (75.0~212.0) | 131.8 (90.2~323.3) | 164.0 (114.0~288.0) |
| 剪接变异 | 7 | 0(0.0) | 2.6 (1.6~7.0) | 736.0 (677.9~1 118.0) | 5 162.0 (1 693.6~5 970.0) | 155.3 (113.0~210.4) | 250.7 (125.0~300.8) | 143.0 (103.0~247.0) |
| 无义变异 | 17 | 0(0.0) | 2.7 (1.3~7.8) | 1 209.0 (589.1~1 442.0) | 6 690.6 (2 783.2~12 500.0) | 118.3 (82.5~311.4) | 200.3 (120.6~422.0) | 117.7 (104.1~253.9) |
| 移码变异 | 6 | 0(0.0) | 1.7 (0.8~3.6) | 9.7 (417.0~1 102.0) | 6 774.0 (1 724.3~7476.7) | 129.6 (114.3~296.0) | 330.0 (191.0~612.2) | 198.0 (126.6~473.7) |
| 统计量 | χ2=2.54 | H=4.54 | H=2.80 | H=4.46 | H=2.78 | H=5.73 | H=7.30 | |
| P | 0.672 | 0.475 | 0.731 | 0.485 | 0.734 | 0.333 | 0.199 |
| [1] | Hoffman EP. Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies[J]. Acta Myol, 2020, 394(4): 179-186. |
| [2] |
Aslesh T, Erkut E, Yokota T. Restoration of dystrophin expression and correction of Duchenne muscular dystrophy by genome editing[J]. Expert Opin Biol Ther, 2021, 21(8): 1049-1061.
doi: 10.1080/14712598.2021.1872539 |
| [3] |
Ma P, Zhang S, Zhang H, et al. Comprehensive genetic characteristics of dystrophinopathies in China[J]. Orphanet J Rare Dis, 2018, 13(1): 109.
doi: 10.1186/s13023-018-0853-z |
| [4] |
Kwon JB, Ettyreddy AR, Vankara A, et al. In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of Duchenne muscular dystrophy[J]. Mol Ther Methods Clin Dev, 2020, 19: 320-329.
doi: 10.1016/j.omtm.2020.09.016 |
| [5] |
Mata López S, Balog-Alvarez C, Vitha S, et al. Correction: challenges associated with homologous directed repair using CRISPR-Cas9 and TALEN to edit the DMD genetic mutation in canine Duchenne muscular dystrophy[J]. PLoS One, 2020, 15(10): e0241430.
doi: 10.1371/journal.pone.0241430 |
| [6] |
Schneider AE, Aartsma-Rus A. Developments in reading frame restoring therapy approaches for Duchenne muscular dystrophy[J]. Expert Opin Biol Ther, 2021, 21(3): 343-359.
doi: 10.1080/14712598.2021.1832462 |
| [7] | 胡亚美, 江载芳, 申昆玲, 等. 诸福棠实用儿科学(下册) [M]. 8版. 北京: 人民卫生出版社, 2015: 2526-2530. |
| [8] |
Richards S, Aziz N, Bale S, et al. standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
doi: 10.1038/gim.2015.30 pmid: 25741868 |
| [9] |
Tyagi R, Arvind H, Goyal M, et al. Working memory alterations plays an essential role in developing global neuropsychological impairment in Duchenne muscular dystrophy[J]. Front Psychol, 2021, 11: 613242.
doi: 10.3389/fpsyg.2020.613242 |
| [10] | Hellebrekers DMJ, Vles JSH, Klinkenberg S, et al. The neurocognitive and behavioral profiles of 3 brothers with Becker muscular dystrophy [J]. Child Neurol Open, 2020, 7: 2329048X20957217. |
| [11] |
Sanchez F, Weitz C, Gutierrez JM, et al. Cardiac MR imaging of muscular dystrophies[J]. Curr Probl Diagn Radiol, 2022, 51(2): 225-234.
doi: 10.1067/j.cpradiol.2020.12.010 |
| [12] |
Lim KRQ, Nguyen Q, Yokota T. Genotype-phenotype correlations in Duchenne and Becker muscular dystrophy patients from the Canadian neuromuscular disease registry[J]. J Pers Med, 2020, 10(4): 241.
doi: 10.3390/jpm10040241 |
| [13] |
Tong YR, Geng C, Guan YZ, et al. A comprehensive analysis of 2013 dystrophinopathies in China: a report from National Rare Disease Center[J]. Front Neurol, 2020, 11: 572006.
doi: 10.3389/fneur.2020.572006 |
| [14] |
Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations[J]. PLoS One, 2015, 10(8): e0135189.
doi: 10.1371/journal.pone.0135189 |
| [15] |
Tawalbeh S, Samsel A, Gordish-Dressman H, et al. Comparison of serum pharmacodynamic biomarkers in prednisone-versus deflazacort-treated Duchenne muscular dystrophy boys[J]. J Pers Med, 2020, 10(4): 164.
doi: 10.3390/jpm10040164 |
| [16] | Merlini L, Cecconi I, Parmeggiani A, et al. Quadriceps muscle strength in Duchenne muscular dystrophy and effect of corticosteroid treatment[J]. Acta Myol, 2020, 39(4): 200-206. |
| [17] |
Sussman MD, Sienko SE, Buckon CE, et al. Efficacy of corticosteroid in decreasing scoliosis and extending time to loss of ambulation in a single clinic: an effectiveness trial[J]. J Child Orthop, 2020, 14(5): 421-432.
doi: 10.1302/1863-2548.14.200156 |
| [18] |
Wood CL, Hollingsworth KG, Hughes E, et al. Pubertal induction in adolescents with DMD is associated with high satisfaction, gonadotropin release and increased muscle contractile surface area[J]. Eur J Endocrinol, 2021, 184(1): 67-79.
doi: 10.1530/EJE-20-0709 |
| [19] |
Panovský R, Pešl M, Máchal J, et al. Quantitative assessment of left ventricular longitudinal function and myocardial deformation in Duchenne muscular dystrophy patients[J]. Orphanet J Rare Dis, 2021, 16(1): 57.
doi: 10.1186/s13023-021-01704-9 pmid: 33516230 |
| [20] |
Mayer OH. The impact of corticosteroids on cardio-pulmonary progression in Duchenne muscular dystrophy[J]. Chest, 2020, 158(5): 1827-1828.
doi: 10.1016/j.chest.2020.05.517 |
| [21] |
Santos ALYDS, Maciel FKL, Fávero FM, et al. Trunk control and upper limb function of walking and non-walking Duchenne muscular dystrophy individuals[J]. Dev Neurorehabil, 2021, 24(7): 435-441.
doi: 10.1080/17518423.2020.1869337 |
| [22] |
Lobo-Prat J. Enkaoua A, Rodriguez-Fernández A, et al. Evaluation of an exercise-enabling control interface for powered wheelchair users: a feasibility study with Duchenne muscular dystrophy[J]. J Neuroeng Rehabil, 2020, 17(1): 142.
doi: 10.1186/s12984-020-00760-9 |
| [23] |
Beckers P, Caberg JH, Dideberg V, et al. Newborn screening of duchenne muscular dystrophy specifically targeting deletions amenable to exon-skipping therapy[J]. Sci Rep, 2021, 11(1): 3011.
doi: 10.1038/s41598-021-82725-z pmid: 33542429 |
| [24] |
Ng MY, Li H, Ghelfi MD, et al. Ataluren and ami-noglycosides stimulate read-through of nonsense codons by orthogonal mechanisms[J]. Proc Natl Acad Sci U S A, 2021, 118(2): e2020599118.
doi: 10.1073/pnas.2020599118 |
| [25] |
Hanson B, Wood MJA, Roberts TC, et al. Molecular correction of Duchenne muscular dystrophy by splice modulation and gene editing[J]. RNA Biol, 2021, 18(7): 1048-1062.
doi: 10.1080/15476286.2021.1874161 |
| [26] |
Anwar S, He M, Lim KRQ, et al. DMDA genotype-phenotype correlation study of exon skip-equivalent in-frame deletions and exon skip-amenable out-of-frame deletions across the DMD gene to simulate the effects of exon-skipping therapies: a meta-analysis[J]. J Pers Med, 2021, 11(1): 46.
doi: 10.3390/jpm11010046 |
| [27] |
Mendell JR, Khan N, Sha N, et al. Comparison of long-term ambulatory function in patients with Duchenne muscular dystrophy treated with eteplirsen and matched natural history controls[J]. J Neuromuscul Dis, 2021, 8(4): 469-479.
doi: 10.3233/JND-200548 pmid: 33523015 |
| [28] |
Scaglioni D, Catapano F, Ellis M, et al. The administration of antisense oligonucleotide golodirsen reduces pathological regeneration in patients with Duchenne muscular dystrophy[J]. Acta Neuropathol Commun, 2021, 9(1): 7.
doi: 10.1186/s40478-020-01106-1 |
| [29] |
Zeng B, Zhou M, Liu B, et al. Targeted addition of mini-dystrophin into rDNA locus of Duchenne muscular dystrophy patient-derived iPSCs[J]. Biochem Biophys Res Commun, 2021, 545: 40-45.
doi: 10.1016/j.bbrc.2021.01.056 |
| [30] |
Sheikh O, Yokota T. Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies[J]. Expert Opin Investig Drugs, 2021, 30(2): 167-176.
doi: 10.1080/13543784.2021.1868434 |
| [1] | Society for Neuroscience and Neurology, Chinese Research Hospital Association, SNN/CRHA. Expert consensus on family care for Duchenne muscular dystrophy (non-ambulatory stage) [J]. Journal of Clinical Pediatrics, 2025, 43(1): 1-7. |
| [2] | CHAI Xingyuan, ZHANG Zhiyong, ZHAO Xiaodong. Progress in radical treatment of X-linked severe combined immunodeficiency [J]. Journal of Clinical Pediatrics, 2024, 42(7): 659-662. |
| [3] | ZHOU Wenhao. The China Neonatal Genome Project: towards a new future in the treatment of genetic diseases [J]. Journal of Clinical Pediatrics, 2024, 42(5): 379-383. |
| [4] | CHEN Guoqing, ZHANG Huiwen. Gene therapy for mucopolysaccharidosis type Ⅱ [J]. Journal of Clinical Pediatrics, 2024, 42(3): 270-276. |
| [5] | LIANG Huan, ZHANG Huiwen. Diagnosis of a child with Duchenne muscular dystrophy using optical genome mapping [J]. Journal of Clinical Pediatrics, 2024, 42(2): 146-150. |
| [6] | Reviewer: DING Yi, YU Yue, Reviser: HAN Lianshu. Research progress in gene therapy for MUT-type methylmalonic acidemia [J]. Journal of Clinical Pediatrics, 2024, 42(12): 1051-1055. |
| [7] | WANG Libo, ZHANG Qianwen, YAO Ruen, TANG Yijun, GAO Shiyang, LI Zhiying, HU Feihan, LI Xin, LOU Dan, WANG Xiumin. Clinical characterization of five children with 17p13.3 microdeletion syndrome and evaluation of their efficacy [J]. Journal of Clinical Pediatrics, 2024, 42(11): 942-947. |
| [8] | JI Taoyun. Prospect of gene therapy for developmental and epileptic encephalopathy [J]. Journal of Clinical Pediatrics, 2023, 41(9): 650-655. |
| [9] | XI Bixin, HU Qun, LIU Aiguo. Advances in the gene therapy for Fanconi anemia [J]. Journal of Clinical Pediatrics, 2023, 41(2): 156-160. |
| [10] | XU Yongli, YANG Jing, ZHOU Lanqi, ZHOU Jianhua. 17q12 microdeletion syndrome: a report of three cases and literature review [J]. Journal of Clinical Pediatrics, 2023, 41(1): 60-65. |
| [11] | WANG Yanyun, SUN Yun, JIANG Tao. 10q22.3-q23.2 deletion syndrome combined with hypermethioninemia: a case report and literature review [J]. Journal of Clinical Pediatrics, 2021, 39(9): 660-. |
| [12] | LIANG Defeng, GENG Lanlan, REN Lu, et al. Endoscopic fenestration for duodenal duplication in children: a case report and literature review [J]. Journal of Clinical Pediatrics, 2021, 39(7): 495-. |
| [13] | CUI Jieyuan, ZHANG Dongfeng, LI Chunzhen, et al. Childhood atypical hemolytic uremic syndrome caused by CFHR family gene variation: a case report and literature review [J]. Journal of Clinical Pediatrics, 2021, 39(2): 95-. |
| [14] | CHEN Jing, TIAN Maoqiang, LI Juan, et al. Nervous system involvement in 2q31.1 microdeletion syndrome caused by mutation in LNPK: a case report and literature review [J]. Journal of Clinical Pediatrics, 2020, 38(6): 459-. |
| [15] | PAN Xiang, LU Jun, LI Guangxu, et al. Chromosome 6p25 deletion syndrome: a case report and literature review [J]. Journal of Clinical Pediatrics, 2020, 38(6): 463-. |
|
||