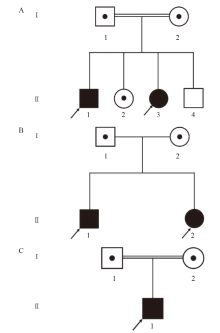
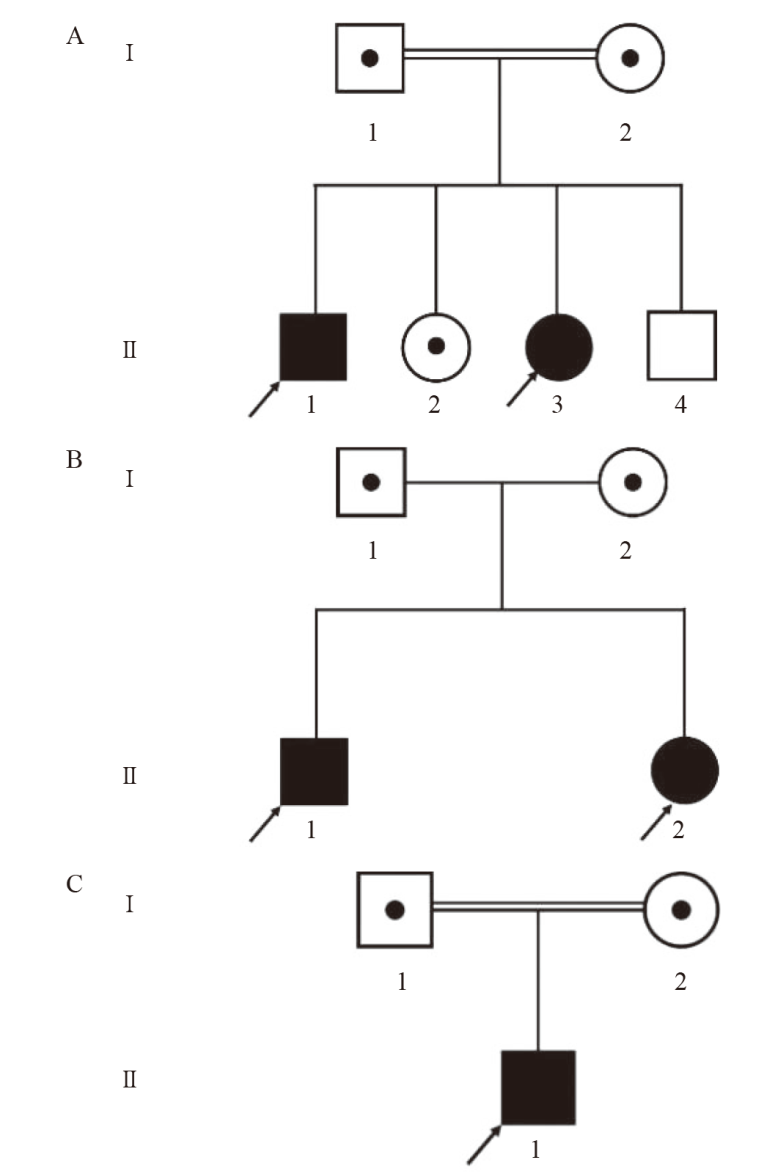
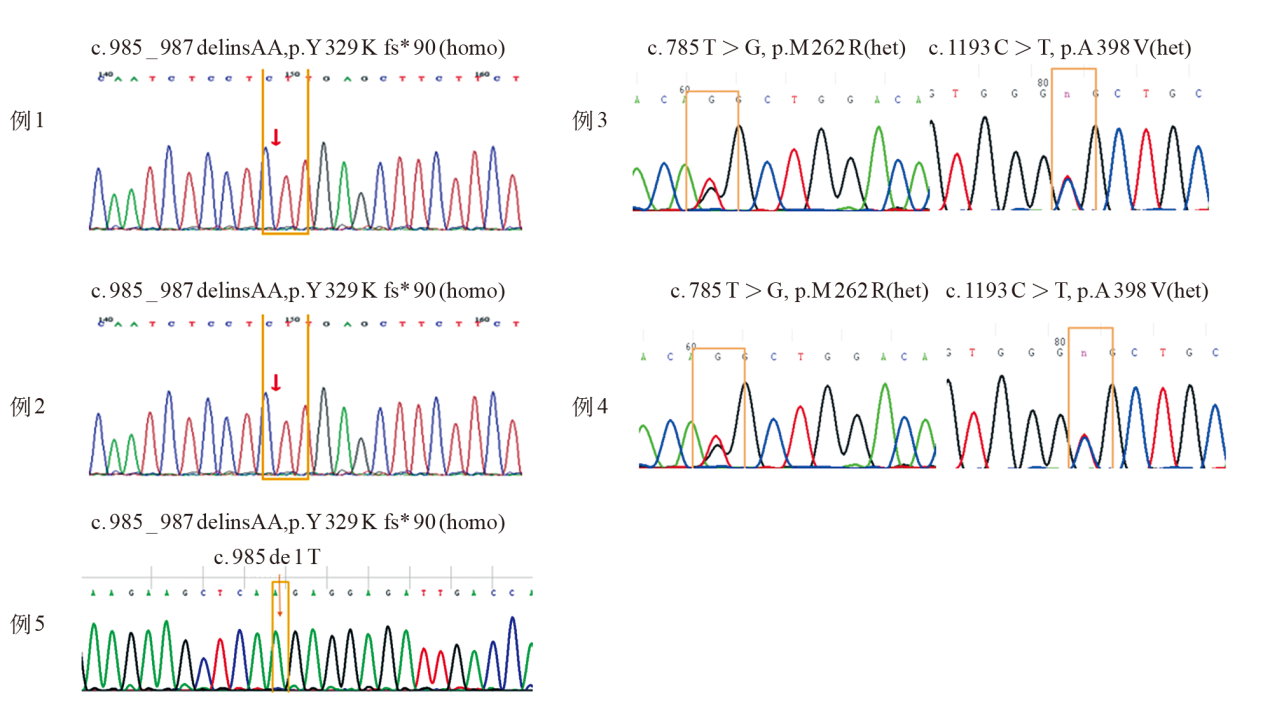
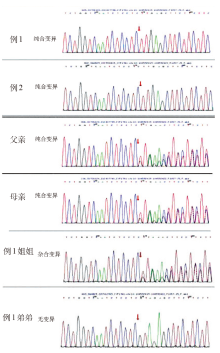
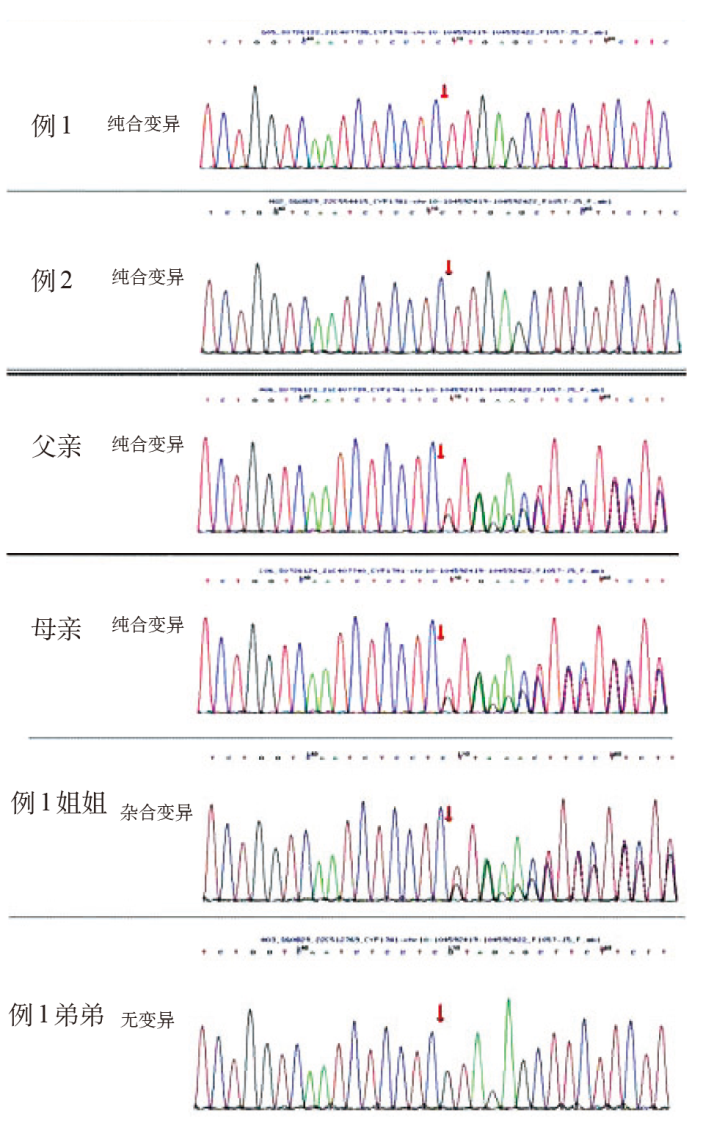
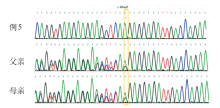
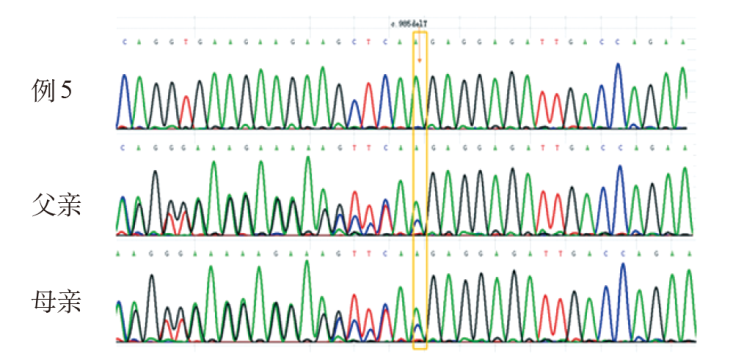
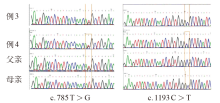
| [1] |
李娟, 王秀敏. 先天性肾上腺皮质增生症的诊治与管理[J]. 中华全科医师杂志, 2023, 22 (6) : 574-579.
|
| [2] |
Kim SM, Rhee JH. A case of 17 alpha-hydroxylase deficiency[J]. Clin Exp Reprod Med, 2015, 42(2): 72-76.
doi: 10.5653/cerm.2015.42.2.72
pmid: 26161337
|
| [3] |
Auchus RJ. The genetics, pathophysiology, and management of human deficiencies of P450c17[J]. Endocrinol Metab Clin North Am, 2001, 30(1): 101-119.
doi: 10.1016/S0889-8529(08)70021-5
|
| [4] |
Hinz L, Pacaud D, Kline G. Congenital adrenal hyperplasia causing hypertension: an illustrative review[J]. J Hum Hypertens, 2018, 32(2): 150-157.
doi: 10.1038/s41371-017-0002-5
pmid: 29255217
|
| [5] |
Bee YM, Manju C, Papari-Zareei M, et al. Phenotypic variation in a Chinese family with 46,XY and 46,XX 17α-hydroxylase deficiency[J]. Gynecol Endocrinol, 2012, 28(4): 322-325.
doi: 10.3109/09513590.2011.631625
pmid: 22087567
|
| [6] |
Martin RM, Lin CJ, Costa EM, et al. P450c17 deficiency in Brazilian patients: biochemical diagnosis through progesterone levels confirmed by CYP17 genotyping[J]. J Clin Endocrinol Metab, 2003, 88(12): 5739-5746.
doi: 10.1210/jc.2003-030988
|
| [7] |
张惠杰, 李小英. 先天性肾上腺增生症与性发育异常[J]. 中国实用内科杂志, 2011, 31(4): 254-256.
|
| [8] |
Chung BC, Picado-Leonard J, Haniu M, et al. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues[J]. Proc Natl Acad Sci U S A, 1987, 84(2): 407-411.
doi: 10.1073/pnas.84.2.407
|
| [9] |
Biglieri EG, Herron MA, Brust N. 17-hydroxylation deficiency in man[J]. J Clin Invest, 1966, 45(12): 1946-1954.
doi: 10.1172/JCI105499
pmid: 4288776
|
| [10] |
胡颖, 时立新, 张淼, 等. 17-a羟化酶/17,20-裂解酶缺乏症1例诊治及相关文献复习[J]. 贵州医药, 2020, 44(3): 459-461.
|
| [11] |
Unal E, Yıldırım R, Taş FF, et al. A rare cause of delayed puberty in two cases with 46,XX and 46,XY karyotype: 17 α-hydroxylase deficiency due to a novel variant in CYP17A1 gene[J]. Gynecol Endocrinol, 2020, 36(8): 739-742.
doi: 10.1080/09513590.2019.1707798
|
| [12] |
Wang M, Wang H, Zhao H, et al. Prevalence of CYP17A1 gene mutations in 17α-hydroxylase deficiency in the Chinese Han population[J]. Clin Hypertens, 2019, 25: 23.
doi: 10.1186/s40885-019-0128-6
|
| [13] |
Hahm JR, Kim DR, Jeong DK, et al. A novel compound heterozygous mutation in the CYP17 (P450 17alpha-hydroxylase) gene leading to 17alpha-hydroxylase/17,20-lyase deficiency[J]. Metabolism, 2003, 52(4): 488-492.
doi: 10.1053/meta.2003.50080
|
| [14] |
张一鸣, 祝捷, 叶山东. 17α-羟化酶/17,20碳链裂解酶缺陷症的研究进展及挑战[J]. 中国临床保健杂志, 2022, 25(2): 285-288.
|
| [15] |
中华医学会儿科学分会内分泌遗传代谢病学组. 先天性肾上腺皮质增生症21-羟化酶缺陷诊治共识[J]. 中华儿科杂志, 2016, 54 (8): 569-576.
|
| [16] |
刘君静, 任丽, 张卫, 等. 成年17α羟化酶缺陷症二例临床特征及诊疗分析[J]. 中国全科医学, 2021, 24(9): 1140-1143.
doi: 10.12114/j.issn.1007-9572.2020.00.571
|
| [17] |
Lees B, Stevenson JC. The prevention of osteoporosis using sequential low-dose hormone replacement therapy with estradiol-17 beta and dydrogesterone[J]. Osteoporos Int, 2001, 12(4): 251-258.
pmid: 11420773
|
| [18] |
蒋敏, 包明晶, 龙恩武, 等. 姐妹同患17-a羟化酶缺乏症病例分析及文献回顾[J]. 中华高血压杂志, 2022, 30(6): 593-597.
|
 ), WANG Xiumin2(
), WANG Xiumin2(