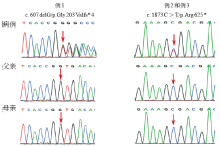
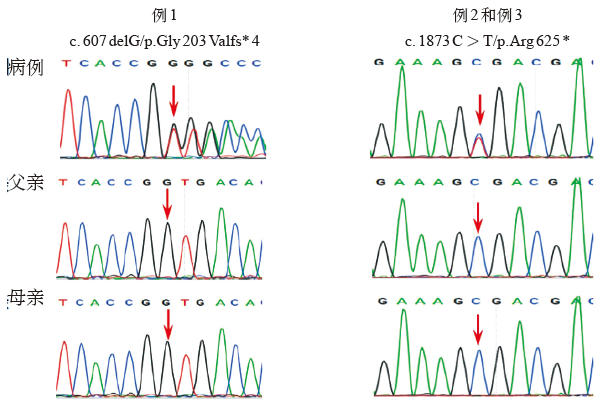
| [1] |
Bellini E, Luoni M, Cancellieri C, et al. SETBP1 accumulation induces P53 inhibition and genotoxic stress in neural progenitors underlying neurodegeneration in Schinzel-Giedion syndrome[J]. Nat Commun, 2021, 12(1): 4050.
doi: 10.1038/s41467-021-24391-3
pmid: 34193871
|
| [2] |
Antonyan L, Ernst C. Putative roles of SETBP1 dosage on the set oncogene to affect brain development[J]. Front Neurosci, 2022, 16: 813430.
|
| [3] |
Jansen NA, Braden RO, Srivastava S, et al. Clinical delineation of SETBP1 haploinsufficiency disorder[J]. Eur J Hum Genet, 2021, 29(8): 1198-1205.
doi: 10.1038/s41431-021-00888-9
pmid: 33867525
|
| [4] |
路通, 王艺. 幼儿特殊面容伴发育落后与多系统畸形[J]. 中国当代儿科杂志, 2017, 19(8): 921-925.
|
| [5] |
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
doi: 10.1038/gim.2015.30
pmid: 25741868
|
| [6] |
Coe BP, Witherspoon K, Rosenfeld JA, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay[J]. Nat Genet, 2014, 46(10): 1063-1071.
doi: 10.1038/ng.3092
pmid: 25217958
|
| [7] |
Morgan A, Braden R, Wong MMK, et al. Speech and language deficits are central to SETBP1 haploinsufficiency disorder[J]. Eur J Hum Genet, 2021, 29(8): 1216-1225.
doi: 10.1038/s41431-021-00894-x
pmid: 33907317
|
| [8] |
Filges I, Shimojima K, Okamoto N, et al. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome[J]. J Med Genet, 2011, 48(2): 117-122.
doi: 10.1136/jmg.2010.084582
|
| [9] |
Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study[J]. Lancet, 2012, 380(9854): 1674-1682.
doi: 10.1016/S0140-6736(12)61480-9
pmid: 23020937
|
| [10] |
Hamdan FF, Srour M, Capo-Chichi JM, et al. De novo mutations in moderate or severe intellectual disability[J]. PLoS Genet, 2014, 10(10): e1004772.
|
| [11] |
Liu WL, He ZX, Li F, et al. Schinzel-Giedion syndrome: a novel case, review and revised diagnostic criteria[J]. J Genet, 2018, 97(1): 35-46.
doi: 10.1007/s12041-017-0877-5
|
| [12] |
Eising E, Carrion-Castillo A, Vino A, et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development[J]. Mol Psychiatry, 2019, 24(7): 1065-1078.
doi: 10.1038/s41380-018-0020-x
|
| [13] |
Leonardi E, Bettella E, Pelizza MF, et al. Identification of SETBP1 mutations by gene panel sequencing in individuals with intellectual disability or with "developmental and epileptic encephalopathy"[J]. Front Neurol, 2020, 11: 593446.
|
| [14] |
Hildebrand MS, Jackson VE, Scerri TS, et al. Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation[J]. Neurology, 2020, 94(20): e2148-e2167.
|
| [15] |
Hu C, Liu D, Luo T, et al. Phenotypic spectrum and long-term outcome of children with genetic early-infantile-onset developmental and epileptic encephalopathy[J]. Epileptic Disord, 2022, 24(2): 343-352.
doi: 10.1684/epd.2021.1394
|
| [16] |
Liu L, Feng X, Liu S, et al. Whole-genome sequencing combined RNA-sequencing analysis of patients with mutations in SET binding protein 1[J]. Front Neurosci, 2022, 16: 980000.
|
 )
)
 沪公网安备 31011002000392号
沪公网安备 31011002000392号