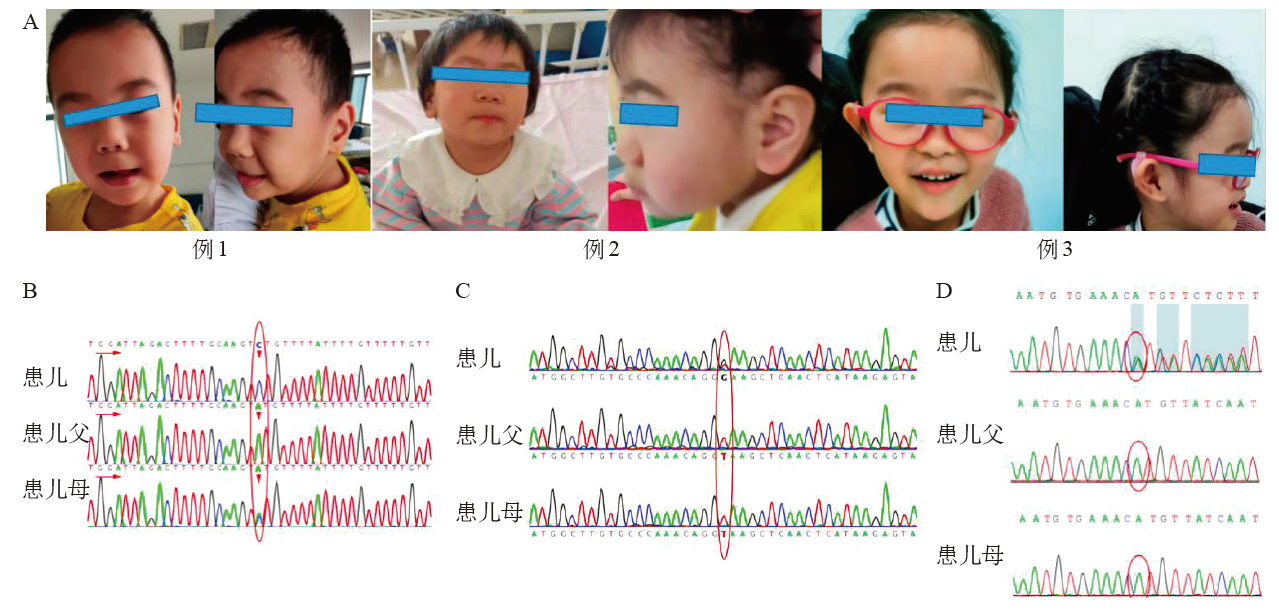
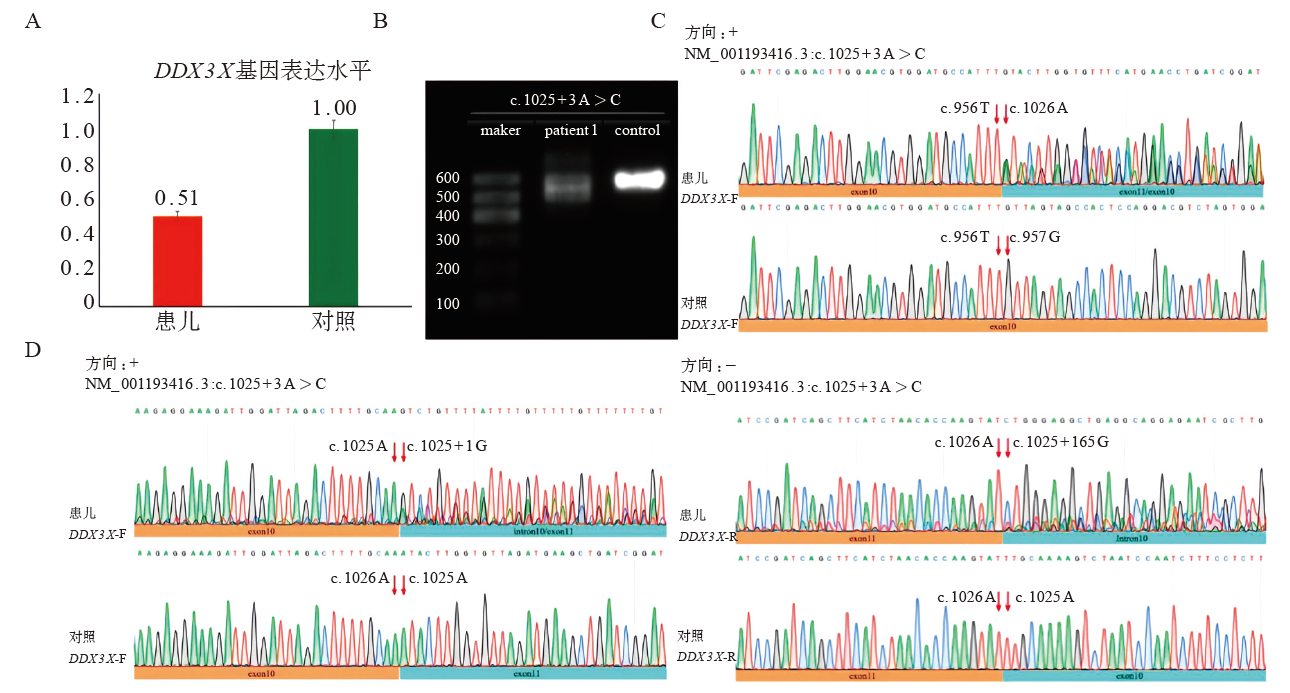
| [1] |
Snijders Blok L, Madsen E, Juusola J, et al. Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling[J]. Am J Hum Genet, 2015, 97(2): 343-352.
doi: 10.1016/j.ajhg.2015.07.004
pmid: 26235985
|
| [2] |
Cruciat CM, Dolde C, de Groot RE, et al. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-β-catenin signaling[J]. Science, 2013, 339(6126): 1436-1441.
|
| [3] |
Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen)[J]. Genet Med, 2020, 22(2): 245-257.
doi: 10.1038/s41436-019-0686-8
pmid: 31690835
|
| [4] |
Dikow N, Granzow M, Graul-Neumann LM, et al. DDX3X mutations in two girls with a phenotype overlapping Toriello-Carey syndrome[J]. Am J Med Genet A, 2017, 173(5): 1369-1373.
|
| [5] |
Fieremans N, Van Esch H, Holvoet M, et al. Identification of intellectual disability genes in female patients with a skewed X-inactivation pattern[J]. Hum Mutat, 2016, 37(8): 804-811.
doi: 10.1002/humu.23012
pmid: 27159028
|
| [6] |
Kellaris G, Khan K, Baig SM, et al. A hypomorphic inherited pathogenic variant in DDX3X causes male intellectual disability with additional neurodevelopmental and neurodegenerative features[J]. Hum Genomics, 2018, 12(1): 11.
|
| [7] |
Nicola P, Blackburn PR, Rasmussen KJ, et al. De novo DDX3X missense variants in males appear viable and contribute to syndromic intellectual disability[J]. Am J Med Genet A, 2019, 179(4): 570-578.
|
| [8] |
Wang X, Posey JE, Rosenfeld JA, et al. Phenotypic expansion in DDX3X - a common cause of intellectual disability in females[J]. Ann Clin Transl Neurol, 2018, 5(10): 1277-1285.
|
| [9] |
Carneiro TN, Krepischi AC, Costa SS, et al. Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases[J]. Appl Clin Genet, 2018, 11: 93-98.
|
| [10] |
Beal B, Hayes I, McGaughran J, et al. Expansion of phenotype of DDX3X syndrome: six new cases[J]. Clin Dysmorphol, 2019, 28(4): 169-174.
|
| [11] |
Scala M, Torella A, Severino M, et al. Three de novo DDX3X variants associated with distinctive brain developmental abnormalities and brain tumor in intellectually disabled females[J]. Eur J Hum Genet, 2019, 27(8): 1254-1259.
|
| [12] |
Chanes NM, Wong J, Lacassie Y. Further delineation of DDX3X syndrome[J]. Clin Dysmorphol, 2019, 28(3): 151-153.
|
| [13] |
Chen Y, Liu KY, Yang ZL, et al. A de novo DDX3X variant is associated with syndromic intellectual disability: case report and literature review[J]. Front Pediatr, 2020, 8: 303.
doi: 10.3389/fped.2020.00303
pmid: 32714884
|
| [14] |
Hu L, Xin X, Lin S, et al. A child with a novel DDX3X variant mimicking cerebral palsy: a case report[J]. Ital J Pediatr, 2020, 46(1): 88.
|
| [15] |
Okano S, Miyamoto A, Makita Y, et al. Severe gastrointestinal symptoms caused by a novel DDX3X variant[J]. Eur J Med Genet, 2020, 63(12): 104058.
|
| [16] |
Lennox AL, Hoye ML, Jiang R, et al. Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development[J]. Neuron, 2020, 106(3): 404-420.
|
| [17] |
Moresco G, Costanza J, Santaniello C, et al. A novel de novo DDX3X missense variant in a female with brachycephaly and intellectual disability: a case report[J]. Ital J Pediatr, 2021, 47(1): 81.
|
| [18] |
Tang L, Levy T, Guillory S, et al. Prospective and detailed behavioral phenotyping in DDX3X syndrome[J]. Mol Autism, 2021, 12(1): 36.
|
| [19] |
高延, 洪琦, 吴慧旦, 等. 基因型-表型关联分析在孤独症谱系障碍早期诊断的研究[J]. 中国儿童保健杂志, 2018, 26(7): 733-736.
doi: 10.11852/zgetbjzz2018-26-07-10
|
| [20] |
刘念, 王越, 史丹丹, 等. DDX3X基因突变致X连锁精神发育迟滞1例并文献复习[J]. 中华实用儿科临床杂志, 2020, 35(15): 1188-1190.
|
| [21] |
王琼, 杨颖, 刘黎礼, 等. 一例DDX3X基因新发变异所致X连锁精神发育迟滞患儿的分析[J]. 中华医学遗传学杂志, 2022, 39(10): 1111-1115.
|
 )
)
 沪公网安备 31011002000392号
沪公网安备 31011002000392号