

Journal of Clinical Pediatrics ›› 2026, Vol. 44 ›› Issue (2): 124-131.doi: 10.12372/jcp.2026.25e1158
• Original Article • Previous Articles Next Articles
HU Xiangsong1,3, LIN Yating2, ZHU Tianwen1( )
)
Received:2025-09-18
Accepted:2025-12-08
Published:2026-02-15
Online:2026-02-02
CLC Number:
HU Xiangsong, LIN Yating, ZHU Tianwen. Natural history of six neonates with severe MTM1-related X-linked myotubular myopathy[J].Journal of Clinical Pediatrics, 2026, 44(2): 124-131.
Table 1
Baseline characteristics and perinatal features of six cases of MTM1-related severe XLMTM"
| 项目 | 例1 | 例2 | 例3 | 例4 | 例5 | 例6 |
|---|---|---|---|---|---|---|
| 性别 | 男 | 男 | 男 | 男 | 男 | 男 |
| 胎龄/周 | 40.86 | 39 | 38.71 | 34.14 | 37.57 | 35 |
| 出生体重/g (百分位数) | 3 050(P6) | 3 800(P85) | 3 170(P41) | 2 310(P57) | 2 500(P8) | 2 090(P13) |
| 出生身长/cm (百分位数) | 50(P22) | 52(P79) | 50(P52) | 45(P49) | 48(P37) | 49(P89) |
| 出生头围/cm (百分位数) | 31(P0) | 36.5(P90) | 34(P31) | 31(P34) | 32(P7) | 32(P41) |
| 母亲年龄/岁 | 34 | 28 | 36 | 35 | 34 | 34 |
| 产前发现 | 孕24周发现球拍状胎盘 | 无 | 无 | 羊水多 | 孕5个月宫颈口松,羊水多 | 羊水多,宫内发育迟缓 |
| 妊娠期疾病 | 无 | 糖尿病 | 甲状腺结节 | 无 | 糖尿病、高血压、GBS阳性 | 甲状腺功能减低 |
| 分娩方式 | 顺产 | 剖宫产 | 剖宫产 | 剖宫产 | 剖宫产 | 剖宫产 |
| Apgar评分 (1-5-10 min) | 3-4-4 | 5-7-7 | 5-6-8 | 6-7-7 | 4-4-6 | 6-7-8 |
| 家族史 | 非近亲婚配;无家族史 | 非近亲婚配;外婆孕育的3名男孩和母亲孕育的2名男孩,均生后数小时死亡,女孩都健康 | 非近亲婚配;无家族史 | 非近亲婚配;前2个男孩,第1个生后因重度窒息死亡,第2个生后肌张力低,生后20天放弃治疗后死亡 | 非近亲婚配;无家族史 | 非近亲婚配;无家族史 |
Table 2
Clinical and examination data for six cases of MTM1-related severe XLMTM"
| 项目 | 例1 | 例2 | 例3 |
|---|---|---|---|
| 起病年龄/d | 0 | 0 | 0 |
| 确诊年龄/d | 37 | 45 | 38 |
| 死亡年龄/d | 3 | 4 | 22 |
| 临床表型 | |||
| 呼吸系统 | 生后自主呼吸弱,气胸 | 生后自主呼吸弱 | 生后自主呼吸弱 |
| 循环系统 | 休克 | 心率慢 | 无影响 |
| 肌肉骨骼系统 | 全身松软 | 肌张力低 | 肌张力低 |
| 外观畸形 | 双手及足细长,阴茎小,隐睾 | 无 | 无 |
| 胸片 | 右侧气胸,两肺炎症 | 无异常 | 无异常 |
| 彩色多普勒超声心动图 | PDA(2 mm双向分流)、ASD(Ⅱ型2.8 mm) | ASD(Ⅱ型4 mm)、PH(44 mmHg) | ASD(Ⅱ型3 mm)、PDA(1.2 mm左向右分流)、PH(30 mmHg) |
| 头颅B超 | 无异常 | 无异常 | 无异常 |
| 头颅CT/MRI | 无异常 | N/A | N/A |
| 脑电图 | 脑电活动低下 | N/A | N/A |
| 代谢检查 | |||
| 血肌酐/μmol·L-1 | 47.0 | 41.6 | 57.0 |
| 丙氨酸氨基转移酶/U·L-1 | 7 | 27 | 30 |
| 血氨/μmol·L-1 | 95 | 52 | N/A |
| 血乳酸/μmol·L-1 | 4.7 | 2.8 | 1.3 |
| 肌酸激酶/U·L-1 | 842 | 732 | N/A |
| 血串联质谱 | 无异常 | 无异常 | 无异常 |
| 呼吸支持 | 有创通气,闭式引流 | 有创通气 | 有创通气改无创 |
| 循环支持 | 肾上腺素、多巴胺 | 多巴胺 | 无 |
| 脑功能支持 | 亚低温 | 无 | 亚低温 |
| 营养支持 | 静脉营养 | 静脉营养 | 静脉营养过渡到鼻饲全肠道喂养 |
| NICU住院时间/d | 3 | 4 | 22 |
| 项目 | 例4 | 例5 | 例6 |
| 起病年龄/d | 0 | 0 | 0 |
| 确诊年龄/d | 50 | 58 | 43 |
| 死亡年龄/d | 150 | 42 | 45 |
| 临床表型 | |||
| 呼吸系统 | 生后自主呼吸弱 | 生后自主呼吸弱 | 生后自主呼吸弱 |
| 循环系统 | 无影响 | 心力衰竭 | 无影响 |
| 肌肉骨骼系统 | 肌张力低 | 全身松软,股骨纤细 | 全身松软 |
| 外观畸形 | 头发细,短绒,隐睾,喉软化 | 无 | 小下颌,被动张嘴困难,隐睾 |
| 胸片 | 反复肺部感染 | 右肺炎症 | 右肺不张 |
| 彩色多普勒超声心动图 | ASD(Ⅱ型4.5 mm) | PDA(2 mm左向右分流)、ASD(Ⅱ型3 mm) | ASD (Ⅱ型2 mm) |
| 头颅B超 | 无异常 | 双侧侧脑室前角及体部扩张 | 双侧脑室周围回声增强 |
| 头颅CT/MRI | N/A | N/A | 蛛网膜下腔少量出血 |
| 脑电图 | N/A | N/A | N/A |
| 代谢检查 | |||
| 血肌酐/μmol·L-1 | 45 | 15.9 | 42 |
| 丙氨酸氨基转移酶/U·L-1 | 32 | 12 | 9 |
| 血氨/μmol·L-1 | 56 | 38 | N/A |
| 血乳酸/μmol·L-1 | 3.1 | 3.4 | 1.3 |
| 肌酸激酶/U·L-1 | 745 | 50 | N/A |
| 血串联质谱 | 无异常 | 无异常 | 无异常 |
| 呼吸支持 | 有创通气改无创 | 有创通气 | 有创通气改无创 |
| 循环支持 | 无 | 肾上腺素、多巴胺 | 无 |
| 脑功能支持 | 无 | 无 | 无 |
| 营养支持 | 静脉营养过渡到鼻饲全肠道喂养 | 静脉营养过渡到鼻饲全肠道喂养 | 静脉营养过渡到鼻饲全肠道喂养 |
| NICU住院时间/d | 52 | 42 | 45 |
Table 3
Genetic variants in six cases of MTM1-related severe XLMTM"
| 患儿 | 核苷酸变化 | 氨基酸变化 | 变异来源 | 是否报道 | ACMG分类 | 结构域位置 |
|---|---|---|---|---|---|---|
| 例1 | Del exon 3 | p.? | 母亲杂合 | 是 | 致病 | RID |
| 例2 | c.614C>T | p.Pro205Leu | 母亲杂合 | 是 | 致病 | RID |
| 例3 | c.591_594del | p.Tyr198Leufs*51 | 母亲杂合 | 是 | 致病 | RID |
| 例4 | Del exon 2 | p.? | 母亲杂合 | 是 | 致病 | RID |
| 例5 | c.1125C>A | p.Cys375* | 母亲杂合 | 否 | 致病 | PTP |
| 例6 | c.614C>T | p.Pro205Leu | 新发变异 | 是 | 致病 | RID |
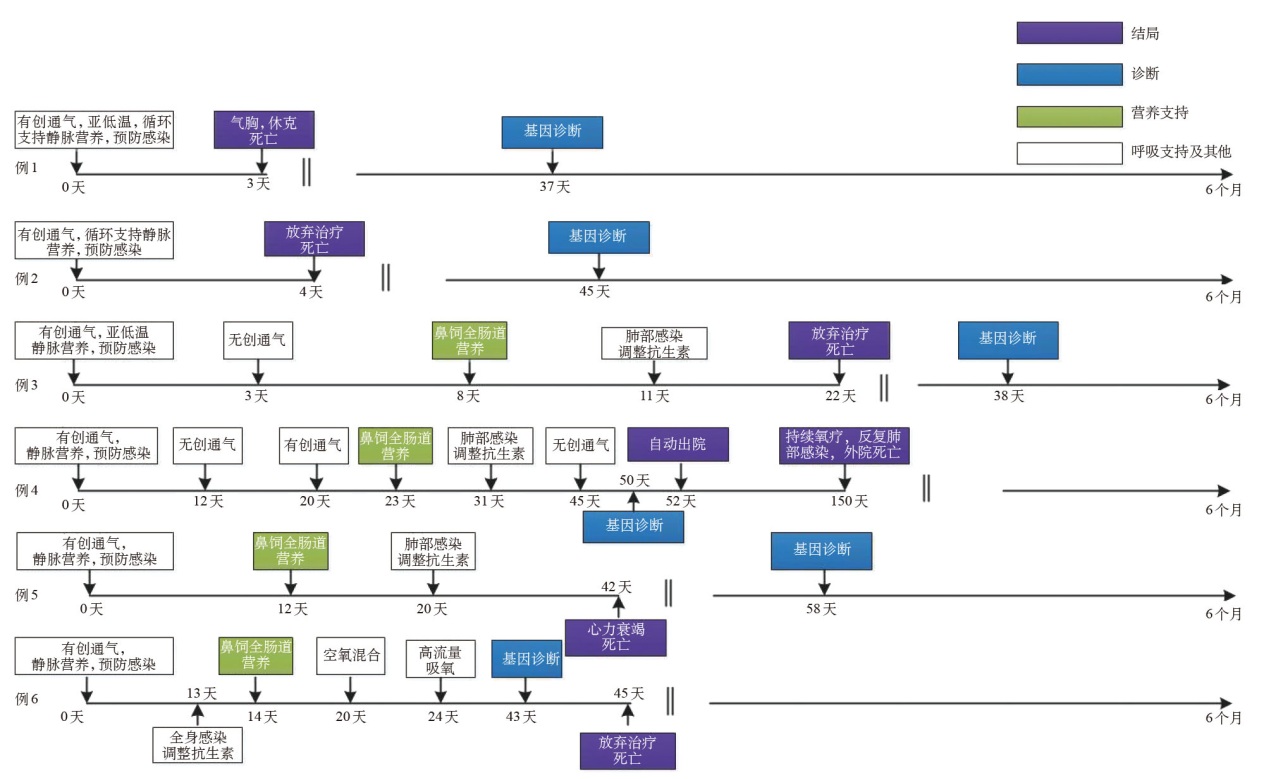
Figure 1
Clinical course of six neonates with MTM1-related severe XLMTM"
| [1] |
Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast[J]. Nat Genet, 1996, 13(2): 175-182.
pmid: 8640223 |
| [2] |
Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy[J]. Orphanet J Rare Dis, 2008, 3: 26.
doi: 10.1186/1750-1172-3-26 pmid: 18817572 |
| [3] |
Lawlor MW, Dowling JJ. X-linked myotubular myopathy[J]. Neuromuscul Disord, 2021, 31(10): 1004-1012.
doi: 10.1016/j.nmd.2021.08.003 |
| [4] |
Beggs AH, Byrne BJ, De Chastonay S, et al. A multicenter, retrospective medical record review of X-linked myotubular myopathy: the recensus study[J]. Muscle Nerve, 2018, 57(4): 550-560.
doi: 10.1002/mus.26018 pmid: 29149770 |
| [5] |
Martin C, Servais L. X-linked myotubular myopathy: an untreated treatable disease[J]. Expert Opin Biol Ther, 2025, 25(4): 379-394.
doi: 10.1080/14712598.2025.2473430 |
| [6] |
Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies[J]. J Child Neurol, 2012, 27(3): 363-382.
doi: 10.1177/0883073812436605 pmid: 22431881 |
| [7] | 胡勇, 黄希. 新发突变基因的新生儿X-连锁肌小管肌病1例并文献回顾[J]. 中南大学学报(医学版), 2024, 49(3): 491-496. |
| Hu Y, Huang X. Neonatal X-linked myotubular myopathy with a de novo mutation: A case report and literature review[J]. Zhongnan Daxue Xuebao (Yixueban), 2024, 49(3): 491-496. | |
| [8] | 付杰, 徐承琴, 郑爱华, 等. MTM1基因半合子突变致新生儿X连锁中央核肌病1例[J]. 浙江医学, 2024, 46(22): 2433-2435. |
| Fu J, Xu CQ, Zheng AH, et al. A case of neonatal X-linked centronuclear myopathy caused by a hemizygous mutation in the MTM1 gene[J]. Zhejiang Yixue, 2024, 46(22): 2433-2435. | |
| [9] | 王钿钿, 王瑜, 王化彬. MTM1基因半合子突变及RYR1基因杂合突变致新生儿X-连锁肌管肌病2例临床分析[J]. 中华实用诊断与治疗杂志, 2023, 37(7): 722-726. |
| Wang TT, Wang Y, Wang HB. Clinical analysis of two cases of neonatal X-linked myotubular myopathy caused by hemizygous mutation of MTM1 gene and heterozygous mutation of RYR1 gene[J]. Zhonghua Shiyong Zhenduan Yu Zhiliao Zazhi, 2023, 37(7): 722-726. | |
| [10] | 洪先慧, 李海英, 吴尤佳. MTM1基因突变致新生儿肌管性肌病1例[J]. 儿科药学杂志, 2023, 29(2): 64-66. |
| Hong XH, Li HY, Wu YJ. A case of neonatal myotubular myopathy caused by MTM1 gene mutation[J]. Erke Yaoxue Zazhi, 2023, 29(2): 64-66. | |
| [11] | 谢添, 葛佳静, 张子明, 等. MTM1基因变异致5例新生儿中央核肌病的临床及遗传学分析[J]. 中国当代儿科杂志, 2025, 27(9): 1071-1075. |
| Xie T, Ge JJ, Zhang ZM, et al. Clinical and genetic features of 5 neonates with centronuclear myopathy caused by MTM1 gene variation[J]. Zhongguo Dangdai Erke Zazhi, 2025, 27(9): 1071-1075. | |
| [12] | Dowling JJ, Lawlor MW, Das S. X-Linked Myotubular Myopathy. 2002 Feb 25 [updated 2018 Aug 23]. In: Adam MP, Bick S, Mirzaa GM, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. |
| [13] |
Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen)[J]. Genet Med, 2020, 22(2): 245-257.
doi: 10.1038/s41436-019-0686-8 pmid: 31690835 |
| [14] |
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
doi: 10.1038/gim.2015.30 pmid: 25741868 |
| [15] | Annoussamy M, Lilien C, Gidaro T, et al. X-linked myotubular myopathy: a prospective international natural history study[J]. Neurology, 2019, 92(16): e1852-e1867. |
| [16] |
Amburgey K, Tsuchiya E, de Chastonay S, et al. A natural history study of X-linked myotubular myopathy[J]. Neurology, 2017, 89(13): 1355-1364.
doi: 10.1212/WNL.0000000000004415 pmid: 28842446 |
| [17] |
Darras BT, De Vivo DC. Precious SMA natural history data: a benchmark to measure future treatment successes[J]. Neurology, 2018, 91(8): 337-339.
doi: 10.1212/WNL.0000000000006026 pmid: 30045956 |
| [18] | Bhattacharyya T, Ghosh A, Verma S, et al. Structural rationale to understand the effect of disease-associated mutations on Myotubularin[J]. Curr Res Struct Biol, 2023, 5: 100100. |
| [19] |
McEntagart M, Parsons G, Buj-Bello A, et al. Genotype-phenotype correlations in X-linked myotubular myopathy[J]. Neuromuscul Disord, 2002, 12(10): 939-946.
doi: 10.1016/S0960-8966(02)00153-0 |
| [20] |
Fattori F, Maggi L, Bruno C, et al. Centronuclear myopathies: genotype-phenotype correlation and frequency of defined genetic forms in an Italian cohort[J]. J Neurol, 2015, 262(7): 1728-1740.
doi: 10.1007/s00415-015-7757-9 pmid: 25957634 |
| [21] |
Shieh PB, Kuntz NL, Dowling JJ, et al. Safety and efficacy of gene replacement therapy for X-linked myotubular myopathy (ASPIRO): a multinational, open-label, dose-escalation trial[J]. Lancet Neurol, 2023, 22(12): 1125-1139.
doi: 10.1016/S1474-4422(23)00313-7 pmid: 37977713 |
| [22] |
Lawlor MW, Schoser B, Margeta M, et al. Effects of gene replacement therapy with resamirigene bilparvovec (AT132) on skeletal muscle pathology in X-linked myotubular myopathy: results from a substudy of the ASPIRO open-label clinical trial[J]. EBioMedicine, 2024, 99: 104894.
doi: 10.1016/j.ebiom.2023.104894 |
| [23] |
Andreoletti G, Romano O, Chou HJ, et al. High-throughput transcriptome analyses from ASPIRO, a phase 1/2/3 study of gene replacement therapy for X-linked myotubular myopathy[J]. Am J Hum Genet, 2023, 110(10): 1648-1660.
doi: 10.1016/j.ajhg.2023.08.008 pmid: 37673065 |
| [24] |
Voermans NC, Ferreiro A, Aartsema-Rus A, et al. Gene therapy for X-linked myotubular myopathy: the challenges[J]. Lancet Neurol, 2023, 22(12): 1089-1091.
doi: 10.1016/S1474-4422(23)00416-7 pmid: 37977700 |
| [25] |
Wilson JM, Flotte TR. Moving forward after two deaths in a gene therapy trial of myotubular myopathy[J]. Hum Gene Ther, 2020, 31(13-14): 695-696.
doi: 10.1089/hum.2020.182 pmid: 32605399 |
| [26] |
Ross LF, Clarke AJ. A historical and current review of newborn screening for neuromuscular disorders from around the world: lessons for the United States[J]. Pediatr Neurol, 2017, 77: 12-22.
doi: S0887-8994(16)30965-1 pmid: 29079012 |
| [27] |
Dangouloff T, Boemer F, Servais L. Newborn screening of neuromuscular diseases[J]. Neuromuscul Disord, 2021, 31(10): 1070-1080.
doi: 10.1016/j.nmd.2021.07.008 |
| [28] | Dangouloff T, Lang H, Benmhammed N, et al. Newborn screening and rapid genomic diagnosis of neuromuscular diseases[J]. J Neuromuscul Dis, 2025, 12(2): 157-172. |
| [29] |
Tanner SM, Laporte J, Guiraud-Chaumeil C, et al. Confirmation of prenatal diagnosis results of X-linked recessive myotubular myopathy by mutational screening, and description of three new mutations in the MTM1 gene[J]. Hum Mutat, 1998, 11(1): 62-68.
pmid: 9450905 |
| [30] |
De Rycke M, Berckmoes V. Preimplantation genetic testing for monogenic disorders[J]. Genes (Basel), 2020, 11(8): 871.
doi: 10.3390/genes11080871 |
| [31] |
Luglio A, Maggi E, Riviello FN, et al. Hereditary neuromuscular disorders in reproductive medicine[J]. Genes (Basel), 2024, 15(11): 1409.
doi: 10.3390/genes15111409 |
| [1] | ZHANG Wenjing, WANG Fan, ZHANG Li, LI Lan. Advances in risk factors and novel biomarkers for neonatal acute kidney injury [J]. Journal of Clinical Pediatrics, 2026, 44(1): 71-78. |
| [2] | HUANG Jing, LIU Xiaochen, GAO Xiong, LI Xiaowen, LI Guanghong, LI Luquan. The value of measuring trace elements and oxidative/antioxidant factors in umbilical blood in predicting the onset and evaluation of neonatal respiratory distress syndrome [J]. Journal of Clinical Pediatrics, 2025, 43(3): 204-210. |
| [3] | HE Ying, LIU Zhiyong, YANG Hansong, CAI Yali, XU Jinglin, CHEN Dongmei. Clinical analysis of 153 neonatal enterovirus infections and antibiotic management improvement study [J]. Journal of Clinical Pediatrics, 2025, 43(2): 128-134. |
| [4] | CHEN Chao. Significance of introducing the concept of “small vulnerable newborns” [J]. Journal of Clinical Pediatrics, 2025, 43(11): 803-806. |
| [5] | LI Jing, LIANG Xuxia, ZHANG Jihong, ZHANG Chun, LU Yanqun, WU Hua. Parental factors associated with singleton small vulnerable neonates: a 5-year retrospective cohort study [J]. Journal of Clinical Pediatrics, 2025, 43(11): 807-815. |
| [6] | ZHENG Ruixue, SUN Xiaodong, WU Guilan, SHEN Leilei. Neonatal refractory congenital chylothorax: two case reports and literature review [J]. Journal of Clinical Pediatrics, 2025, 43(10): 775-781. |
| [7] | ZHANG Ruijie, XI Guannan, WANG Xuefeng, LIN Xin’ao, DAI Jiale, FAN Xiaobo, WANG Jimei. Application of simplified lung ultrasound performed shortly after birth in the respiratory support needs of late preterm and term infants [J]. Journal of Clinical Pediatrics, 2024, 42(8): 714-721. |
| [8] | WANG Man, LI Luquan, LI Xiaowen. Analysis of risk factors for early-onset sepsis associated acute kidney injury in neonates [J]. Journal of Clinical Pediatrics, 2024, 42(6): 520-525. |
| [9] | XIANG Chao, ZHANG Rong, KANG Lan, LEI Xiaoping, LIU Xingqing, DONG Wenbin. Association of coefficient of glycemic variation and SNAPPE-Ⅱ with prognosis in critically ill neonates [J]. Journal of Clinical Pediatrics, 2023, 41(6): 430-435. |
| [10] | XU Jinglin, YANG Hansong, CHEN Xinhua, CHEN Jiangbin, LI Xiaoqing, ZHANG Weifeng, CHEN Dongmei. Clinical analysis of continuous blood purification in the treatment of neonatal septic shock with acute kidney injury [J]. Journal of Clinical Pediatrics, 2023, 41(6): 436-441. |
| [11] | ZHANG Yongjun, ZHU Tianwen. Early diagnosis and precise intervention of neonatal hyperammonemia [J]. Journal of Clinical Pediatrics, 2023, 41(4): 241-246. |
| [12] | Shenzhen Neonatal Data Network. A multicenter survey and clinical analysis of neonatal hyperammonemia [J]. Journal of Clinical Pediatrics, 2023, 41(4): 252-258. |
| [13] | CHU Xiaoyun, SUN Yifan, YAN Chongbing, HONG Wenchao, GONG Xiaohui, CAI Cheng. Clinical analysis of urea cycle disorders in 5 neonates [J]. Journal of Clinical Pediatrics, 2023, 41(4): 266-271. |
| [14] | HU Xuyun, HAO Chanjuan. Genetic diagnosis and management of TRPV4 disorders [J]. Journal of Clinical Pediatrics, 2023, 41(2): 86-91. |
| [15] | LIU Ling, JIANG Yuhui, NIE Panrong, ZENG Limei, DUAN Gaiyuan, LI Yuchen. Investigation of the relationship between gene polymorphisms and neonatal hyperbilirubinemia in southwest China [J]. Journal of Clinical Pediatrics, 2022, 40(9): 672-678. |
|
||