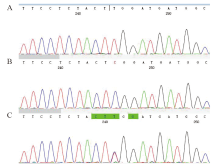
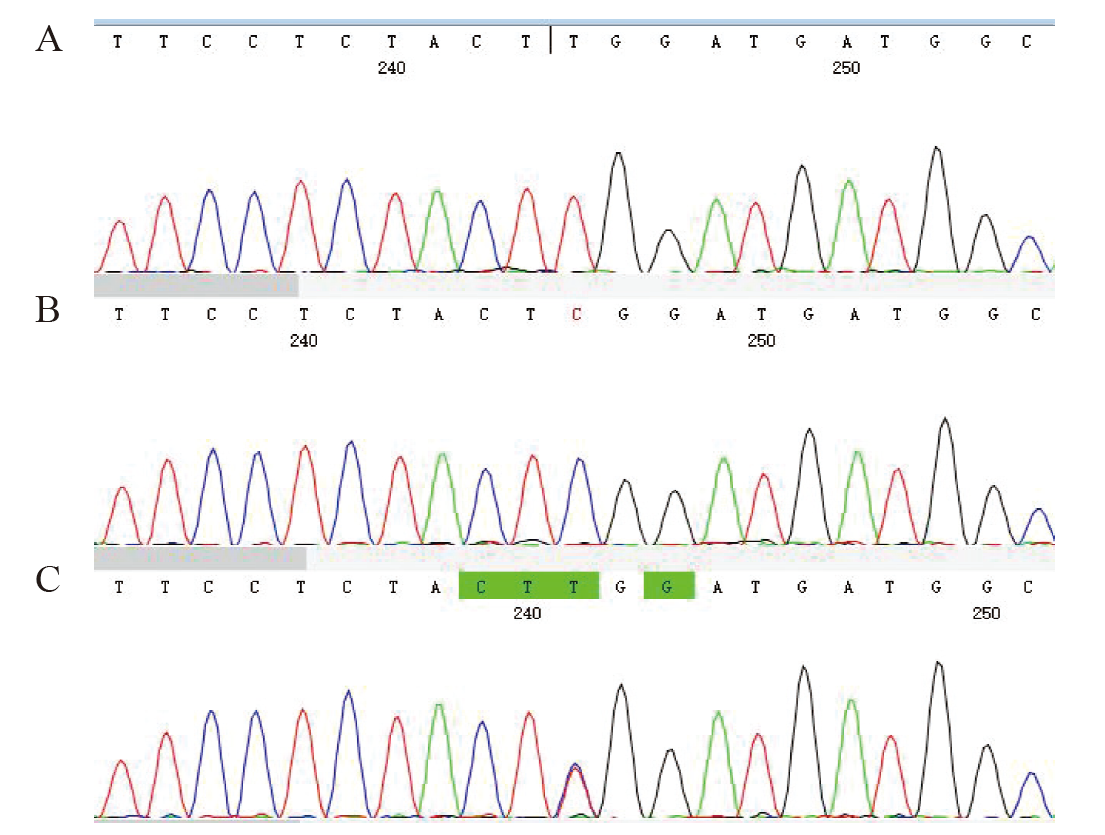
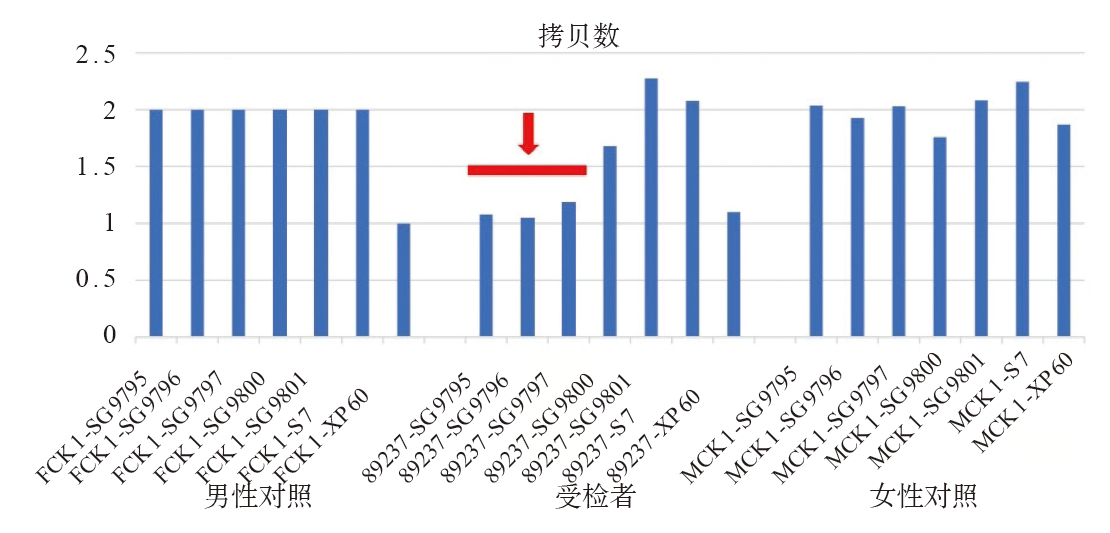
| [1] |
Fischer MH, Gerritsen T, Opitz JM. Alpha-aminoadipic aciduria, a non-deleterious inborn metabolic defect[J]. Humangenetik, 1974, 24(4): 265-270.
pmid: 4442872
|
| [2] |
Stiles AR, Venturoni L, Mucci G, et al. New cases of DHTKD1 mutations in patients with 2-ketoadipic aciduria[J]. JIMD Rep, 2016, 25: 15-19.
|
| [3] |
Leandro J, Violante S, Argmann CA, et al. Mild inborn errors of metabolism in commonly used inbred mouse strains[J]. Mol Genet Metab, 2019, 126(4): 388-396.
doi: S1096-7192(18)30716-9
pmid: 30709776
|
| [4] |
Nemeria NS, Gerfen G, Yang L, et al. Evidence for functional and regulatory cross-talk between the tricarboxylic acid cycle 2-oxoglutarate dehydrogenase complex and 2-oxoadipate dehydrogenase on the l-lysine, l-hydroxylysine and l-tryptophan degradation pathways from studies in vitro[J]. Biochim Biophys Acta Bioenerg, 2018, 1859(9): 932-939.
doi: 10.1016/j.bbabio.2018.05.001
|
| [5] |
Leandro J, Houten SM. The lysine degradation pathway: subcellular compartmentalization and enzyme deficiencies[J]. Mol Genet Metab, 2020, 131(1-2): 14-22.
doi: 10.1016/j.ymgme.2020.07.010
pmid: 32768327
|
| [6] |
Hagen J, Te Brinke H, Wanders RJ, et al. Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria[J]. J Inherit Metab Dis, 2015, 38(5): 873-879.
doi: 10.1007/s10545-015-9841-9
pmid: 25860818
|
| [7] |
da Silva JC, Amaral AU, Cecatto C, et al. α-ketoadipic acid and α-aminoadipic acid cause disturbance of glutamatergic neurotransmission and induction of oxidative stress in vitro in brain of adolescent rats[J]. Neurotox Res, 2017, 32(2): 276-290.
doi: 10.1007/s12640-017-9735-8
pmid: 28429309
|
| [8] |
Rodenburg RJ. The functional genomics laboratory: functional validation of genetic variants[J]. J Inherit Metab Dis, 2018, 41(3): 297-307.
doi: 10.1007/s10545-018-0146-7
pmid: 29445992
|
| [9] |
Duran M, Beemer FA, Wadman SK, et al. A patient with α-ketoadipic and α-aminoadipic aciduria[J]. J Inherit Metab Dis, 1984, 7(2): 61.
pmid: 6434826
|
| [10] |
Xia Z W, Inoue Y, Ohse M, et al. A study on α-ketoadipic aciduria by gas chromatographic-mass spectrometry[J]. World Gastroenterol, 2000, 6(5): 766-769.
doi: 10.3748/wjg.v6.i5.766
|
| [11] |
Leandro J, Dodatko T, DeVita RJ, et al. Deletion of 2-aminoadipic semialdehyde synthase limits metabolite accumulation in cell and mouse models for glutaric aciduria type 1[J]. J Inherit Metab Dis, 2020, 43(6): 1154-1164.
doi: 10.1002/jimd.v43.6
|
 )
)