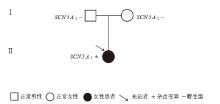
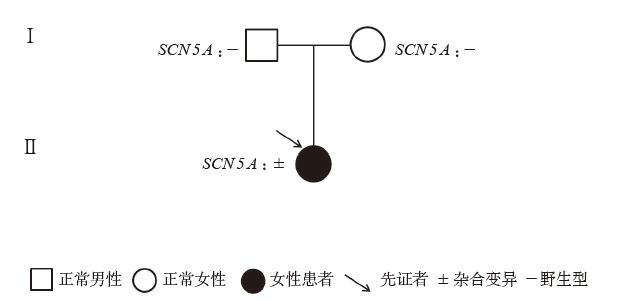
| [1] |
Steinberg C. Diagnosis and clinical management of long-QT syndrome[J]. Curr Opin Cardiol, 2018, 33(1): 31-41.
doi: 10.1097/HCO.0000000000000465
pmid: 28984650
|
| [2] |
Neira V, Enriquez A, Simpson C, et al. Update on long QT syndrome[J]. J Cardiovasc Electrophysiol, 2019, 30(12): 3068-3078.
|
| [3] |
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013[J]. Heart Rhythm, 2013, 10(12): 1932-1963.
doi: 10.1016/j.hrthm.2013.05.014
pmid: 24011539
|
| [4] |
Westphal DS, Burkard T, Moscu-Gregor A, et al. Reclassification of genetic variants in children with long QT syndrome[J]. Mol Genet Genomic Med, 2020, 8(9): e1300.
|
| [5] |
Hofman N, Jongbloed R, Postema PG, et al. Recurrent and founder mutations in the Netherlands: the long-QT syndrome[J]. Neth Heart J, 2011, 19(1): 10-16.
doi: 10.1007/s12471-010-0046-4
pmid: 21350584
|
| [6] |
Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test[J]. Heart Rhythm, 2009, 6(9): 1297-1303.
doi: 10.1016/j.hrthm.2009.05.021
pmid: 19716085
|
| [7] |
Marschall C, Moscu-Gregor A, Klein HG. Variant panorama in 1,385 index patients and sensitivity of expanded next-generation sequencing panels in arrhythmogenic disorders[J]. Cardiovasc Diagn Ther, 2019, 9(Suppl 2): S292-S298.
|
| [8] |
Ng CA, Perry MD, Liang W, et al. High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants[J]. Heart Rhythm, 2020, 17(3): 492-500.
|
| [9] |
Fazelifar AF, Pourirahim M, Masoumi T, et al. Identification of a novel pathogenic variant in KCNH2 in an Iranian family with long QT syndrome 2 by whole-exome sequencing[J]. J Arrhythm, 2023, 39(3): 430-453.
doi: 10.1002/joa3.12857
pmid: 37324772
|
| [10] |
Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing[J]. Heart Rhythm, 2010, 7(1): 33-46.
doi: 10.1016/j.hrthm.2009.09.069
pmid: 20129283
|
| [11] |
Fukuyama M, Ohno S, Ozawa J, et al. High prevalence of late-appearing T-wave in patients with long QT syndrome type 8[J]. Circ J, 2020, 84(4): 559-568.
doi: 10.1253/circj.CJ-19-1101
pmid: 32161207
|
| [12] |
Sun Q, Xie Z, Wang F, et al. Case report of a child with long QT syndrome type 14 caused by CALM1 gene mutation and literature review[J]. Mol Genet Genomic Med, 2024, 12(1): e2287.
|
| [13] |
Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death[J]. Eur Heart J, 2022, 43(40): 3997-4126.
doi: 10.1093/eurheartj/ehac262
pmid: 36017572
|
| [14] |
Barsheshet A, Dotsenko O, Goldenberg I. Congenital long QT syndromes: prevalence, pathophysiology and management[J]. Paediatr Drugs, 2014, 16 (6): 447-456.
doi: 10.1007/s40272-014-0090-4
pmid: 25288402
|
| [15] |
Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy[J]. Heart Rhythm, 2019, 16(11):e301-e372.
|
| [16] |
Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes[J]. Transl Res, 2013, 161(1): 1-14.
doi: 10.1016/j.trsl.2012.08.005
pmid: 22995932
|
| [17] |
Huang H, Kuenze G, Smith JA, et al. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations[J]. Sci Adv, 2018, 4(3): eaar2631.
|
| [18] |
Lahtinen AM, Marjamaa A, Swan H, Kontula K. KCNE1 D85N polymorphism--a sex-specific modifier in type 1 long QT syndrome?[J]. BMC Med Genet, 2011, 12: 11.
doi: 10.1186/1471-2350-12-11
pmid: 21244686
|
| [19] |
Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome[J]. N Engl J Med, 2003, 348(19): 1866-1874.
|
| [20] |
Barsheshet A, Goldenberg I, J OU, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to β-blocker therapy in type 1 long-QT syndrome[J]. Circulation, 2012, 125(16): 1988-1996.
doi: 10.1161/CIRCULATIONAHA.111.048041
pmid: 22456477
|
| [21] |
Copier JS, Bootsma M, Ng CA, et al. Reclassification of a likely pathogenic Dutch founder variant in KCNH2; implications of reduced penetrance[J]. Hum Mol Genet, 2023, 32(7): 1072-1082.
|
| [22] |
Tester DJ, Will ML, Haglund CM, et al. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing[J]. Heart Rhythm, 2005, 2(5): 507-517.
doi: 10.1016/j.hrthm.2005.01.020
pmid: 15840476
|
| [23] |
中华医学会心血管病学分会精准心血管病学学组, 中国医疗保健国际交流促进会精准心血管病分会, 中华心血管病杂志编辑委员会. 单基因遗传性心血管疾病基因诊断指南[J]. 中华心血管病杂志, 2019, 47(3): 175-196.
|
|
Section of Precision Cardiovascular Medicine of Chinese Society of Cardiology; Precision Cardiovascular Medicine Branch of China International Exchange; Promotive Association for Medical; Health Care Editorial Board of Chinese Journal of Cardiology. Guideline for the genetic diagnosis of monogenic cardiovascular diseases[J]. Zhonghua Xinxueguanbing Zazhi, 2019, 47(3): 175-196.
|
| [24] |
中国生物医学工程学会心律分会. 中国儿童心血管植入性电子器械专家共识[J]. 中国心脏起搏与心电生理杂志, 2023, 37(1): 1-11.
|
| [25] |
Rohatgi RK, Sugrue A, Bos JM, et al. Contemporary outcomes in patients with long QT syndrome[J]. J Am Coll Cardiol, 2017, 70(4): 453-462.
doi: S0735-1097(17)37519-8
pmid: 28728690
|
 ), 李东晓3
), 李东晓3
 沪公网安备 31011002000392号
沪公网安备 31011002000392号