

Journal of Clinical Pediatrics ›› 2026, Vol. 44 ›› Issue (2): 139-145.doi: 10.12372/jcp.2026.25e0120
• Original Article • Previous Articles Next Articles
LI Huarong, CHEN Chaoying( ), TU Juan, LIN Tiantian, WANG Nannan
), TU Juan, LIN Tiantian, WANG Nannan
Received:2025-02-17
Accepted:2025-07-14
Published:2026-02-15
Online:2026-02-02
CLC Number:
LI Huarong, CHEN Chaoying, TU Juan, LIN Tiantian, WANG Nannan. Four cases of C3 glomerulopathy in children and literature review[J].Journal of Clinical Pediatrics, 2026, 44(2): 139-145.
Table 1
Clinical characteristics of 4 children with C3G in our center"
| 项目 | 例1 | 例2 | 例3 | 例4 |
|---|---|---|---|---|
| 发病年龄/岁 | 10 | 12 | 12 | 10 |
| 性别 | 女 | 男 | 男 | 女 |
| 浮肿 | + | + | + | + |
| 肉眼血尿 | + | - | + | - |
| 高血压 | + | + | + | + |
| 贫血 | + | + | + | - |
| ALB/g·L-1 | 21.1 | 23 | 29 | 25.5 |
| CHOL/mmol·L-1 | 5.84 | 5.46 | 5.94 | 10.26 |
| Scr/µmol·L-1 | 66.4 | 65 | 101 | 45.5 |
| Urea/mmol·L-1 | 10.7 | 9.83 | 10.2 | 4.6 |
| eGFR/mL·min-1·1.73m-2 | 75.8 | 89 | 59 | 118 |
| UTP/mg·kg-1·d-1 | 269 | 95.7 | 106 | 118.3 |
| UPCR/mg·mg-1 | 10 | 1.11 | 6.59 | 4.82 |
| ASO | - | - | + | - |
| 补体C3/g·L-1 | <0.05 | 0.047 | 0.15 | 0.07 |
| 补体C4/g·L-1 | 0.08 | 0.084 | 0.26 | 0.17 |
| C5b-9/ng·mL-1 | 816 | 2 061 | 924 | 1 748 |
| 肾外表现 | 免疫性 溶血 | 无 | 无 | 双眼屈光 -不正 |
Table 2
Pathological characteristics of 4 children with C3G"
| 项目 | 例1 | 例2 | 例3 | 例4 |
|---|---|---|---|---|
| 发病至肾活检时间/d | 20 | 40 | 25 | 10 |
| 弥漫系膜增生 | + | + | + | + |
| 内皮细胞增生 | - | 局灶节段增生 | 弥漫节段增生 | - |
| 基底膜 | 弥漫增厚,伴系膜插入和双轨形成 | 弥漫增厚,伴系膜插入和双轨形成 | 系膜插入和双轨形成 | 弥漫增厚,伴系膜插入和双轨形成 |
| 白细胞浸润 | - | 少量 | 较多 | 较多 |
| 新月体/% | 0 | 0 | 21.40 | 0 |
| 肾小球硬化/% | 0 | 17.80 | 0 | 3.20 |
| 肾小管间质 | 颗粒及空泡变性,少量红细胞管型,部分刷状缘脱落 | 颗粒及空泡变性,轻度TBM增厚,间质轻度纤维化 | 颗粒变性,少量红细胞管型,散在刷状缘脱落 | 颗粒及空泡变性 |
| 免疫荧光 | ||||
| C3 | ++~+++ | ++ | ++ | +++ |
| C4 | + | - | - | + |
| IgG | ±~+ | - | - | - |
| IgM | + | ± | - | ± |
| IgA | - | - | - | - |
| 电镜 | ||||
| IC沉积部位 | 基底膜 | 系膜区、内皮下、上皮下、基底膜 | 系膜区、内皮下、上皮下、基底膜 | 系膜区、内皮下、上皮下 |
| 足细胞 | 节段融合脱落 | 广泛融合 | 广泛融合 | 广泛融合 |
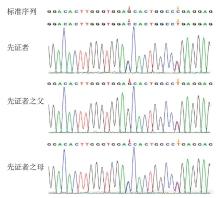
Figure 1
Sanger sequencing results of C3G Case 4 and her parents"
Table 3
Treatment and follow-up of 4 children with C3G"
| 项目 | 例1 | 例2 | 例3 | 例4 |
|---|---|---|---|---|
| 治疗 | ||||
| ACEI | + | + | - | + |
| 糖皮质激素 | + | + | + | + |
| CYC | + | - | - | - |
| MMF | - | + | - | + |
| ECU | + | - | - | + |
| 其他 | PI2次、PE2次 | - | - | - |
| 随访时间/月 | 3 | 6 | 4 | 8 |
| 末次C3/g·L-1 | 0.1 | 0.07 | 0.85 | 0.19 |
| 末次UPCR/mg·mg-1 | 12 | 0.30 | 0.11 | 1.01 |
| 末次UTP/mg·d-1 | 2 232 | 594 | 128 | 1 031 |
| 末次Scr/µmol·L-1 | 35.8 | 54 | 47.2 | 46.2 |
| 末次eGFR/ mL·min-1·1.73m-2 | 140 | 107 | 121 | 118.9 |
| [1] |
Riedl Khursigara M, Matsuda-Abedini M, Radhakrishnan S, et al. A guide for adult nephrologists and hematologists to managing atypical hemolytic uremic syndrome and C3 glomerulopathy in teens transitioning to young adults[J]. Adv Chronic Kidney Dis, 2022, 29(3): 231-242.
doi: 10.1053/j.ackd.2022.04.003 |
| [2] |
Pickering MC, D'Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report[J]. Kidney Int, 2013, 84(6): 1079-1089.
doi: 10.1038/ki.2013.377 pmid: 24172683 |
| [3] |
Cappoli A, Kersnik-Levart T, Silecchia V, et al. C3 glomerulopathy in children: a European longitudinal study evaluating outcome[J]. Pediatr Nephrol, 2025, 40(4): 979-986.
doi: 10.1007/s00467-024-06587-z pmid: 39589481 |
| [4] |
Vivarelli M, van de Kar N, Labbadia R, et al. A clinical approach to children with C3 glomerulopathy[J]. Pediatr Nephrol, 2022, 37(3): 521-535.
doi: 10.1007/s00467-021-05088-7 |
| [5] | 王培培, 张沛, 高春林, 等. 补体参与儿童肾脏疾病发病机制的再认识[J]. 临床儿科杂志, 2024, 42(5): 467-473. |
| Wang PP, Zhang P, Gao CL, et al. Re-recognition of the complement involvement in the pathogenesis of kidney diseases in children[J]. Linchuang Erke Zazhi, 2024, 42(5): 467-473. | |
| [6] | 何瑞娟, 肖慧捷, 王素霞, 等. 伴有H因子下降的儿童C3肾小球病的临床病理三例[J]. 中华儿科杂志, 2012, 50(12): 939-943. |
| He RJ, Xiao HJ, Wang SX, et al. Characteristics of pediatric C3 glomerulopathy with decreased factor H in 3 cases[J]. Zhonghua Erke Zazhi, 2012, 50(12): 939-943. | |
| [7] | 汪瑛, 林强, 朱赟, 等. 儿童C3肾小球肾炎7例[J]. 中华实用儿科临床杂志, 2013, 28(5): 349-351. |
| Wand Y, Lin Q, Zhu Y, et al. C 3 glomerulonephritis in 7 children[J]. Zhonghua Shiyong Erke Linchuang Zazhi, 2013, 28(5): 349-351. | |
| [8] | 霍华丽, 席微波, 曹璐, 等. 儿童C3肾小球肾炎并Alport综合征1例[J]. 中国临床案例成果数据库, 2022, 4(1): E06038-E06038. |
| Huo HL, Xi WB, Cao L, et al. A case report of C3 glomerulonephritis with Alport syndrome in children[J]. Zhongguo Linchuang Anli Chengguo Shujuku, 2022, 4(1): E06038-E06038. | |
| [9] | 杨帆, 邓泂, 陈泽林, 等. 儿童C3肾小球肾炎及其文献复习[J]. 中华妇幼临床医学杂志(电子版), 2017, 13(5): 574-579. |
| Yang F, Deng J, Chen ZL, et al. C3 glomerulonephritis of children and literatures review[J]. Zhonghua Fuyou Linchuang Yixue Zazhi (Dianziban), 2017, 13(5): 574-579. | |
| [10] | 王坤, 吴滢, 王平, 等. 抗H因子抗体阳性儿童致密物沉积病1例[J]. 中华肾脏病杂志, 2022, 38(8): 722-725. |
| Wang K, Wu Y, Wang P, et al. A case report of dense deposit disease with positive anti-factor H antibody in a child[J]. Zhonghua Shenzangbing Zazhi, 2022, 38(8): 722-725. | |
| [11] |
Wei L, Fang Y, Cao G, et al. Genetic and pathological findings in a boy with psoriasis and C3 glomerulonephritis: A case report and literature review[J]. Mol Genet Genomic Med, 2020, 8(10): e1430.
doi: 10.1002/mgg3.v8.10 |
| [12] |
Khandelwal P, Bhardwaj S, Singh G, et al. Therapy and outcomes of C3 glomerulopathy and immune-complex membranoproliferative glomerulonephritis[J]. Pediatr Nephrol, 2021, 36(3): 591-600.
doi: 10.1007/s00467-020-04736-8 |
| [13] |
Günay N, Dursun İ, Gökçe İ, et al. Complement gene mutations in children with C3 glomerulopathy: do they affect the response to mycophenolate mofetil?[J]. Pediatr Nephrol, 2024, 39(5): 1435-1446.
doi: 10.1007/s00467-023-06231-2 |
| [14] |
Zahir Z, Wani AS, Gupta A, et al. Pediatric C3 glomerulopathy: a 12-year single-center experience[J]. Pediatr Nephrol, 2021, 36(3): 601-610.
doi: 10.1007/s00467-020-04768-0 |
| [15] |
Ueda C, Horinouchi T, Inoki Y, et al. Clinical characteristics and outcomes of immune-complex membranoproliferative glomerulonephritis and C3 glomerulopathy in Japanese children[J]. Pediatr Nephrol, 2024, 39(9): 2679-2689.
doi: 10.1007/s00467-024-06377-7 pmid: 38662234 |
| [16] |
Asano M, Oda T, Mizuno M. A case of C3 glomerulopathy with nephritis-associated plasmin receptor positivity without a history of streptococcal infection[J]. CEN Case Rep, 2022, 11(2): 259-264.
doi: 10.1007/s13730-021-00662-2 |
| [17] | 雷蕾, 陈植, 刘小荣. 儿童急性链球菌感染后肾小球肾炎与C3肾小球病的临床分析[J]. 中华实用儿科临床杂志, 2022, 37(21): 1660-1664. |
| Lei L, Chen Z, Liu XR. Clinical analysis of acute post-streptococcal glomerulonephritis and C3 glomerulopathy in children[J]. Zhonghua Shiyong Erke Linchuang Zazhi, 2022, 37(21): 1660-1664. | |
| [18] |
Caravaca-Fontán F, Lucientes L, Cavero T, et al. Update on C3 Glomerulopathy: a complement-mediated disease[J]. Nephron, 2020, 144(6): 272-280.
doi: 10.1159/000507254 |
| [19] | Mehdi A, Taliercio JJ. C3 glomerulopathy[J]. Cleve Clin J Med, 2023, 90(6suppl 1): e1-e4. |
| [20] |
Kirpalani A, Jawa N, Smoyer WE, et al. Long-term outcomes of C3 glomerulopathy and immune-complex membranoproliferative glomerulonephritis in children[J]. Kidney Int Rep, 2020, 5(12): 2313-2324.
doi: 10.1016/j.ekir.2020.09.019 pmid: 33305125 |
| [21] |
Fakhouri F, Le Quintrec M, Frémeaux-Bacchi V. Practical management of C3 glomerulopathy and Ig-mediated MPGN: facts and uncertainties[J]. Kidney Int, 2020, 98(5): 1135-1148.
doi: 10.1016/j.kint.2020.05.053 pmid: 32622830 |
| [22] |
Caravaca-Fontán F, Díaz-Encarnación M, Cabello V, et al. Longitudinal change in proteinuria and kidney outcomes in C3 glomerulopathy[J]. Nephrol Dial Transplant, 2022, 37(7): 1270-1280.
doi: 10.1093/ndt/gfab075 |
| [23] |
Chauvet S, Hauer JJ, Petitprez F, et al. Results from a nationwide retrospective cohort measure the impact of C3 and soluble C5b-9 levels on kidney outcomes in C3 glomerulopathy[J]. Kidney Int, 2022, 102(4):904-916.
doi: 10.1016/j.kint.2022.05.027 pmid: 35752323 |
| [24] | Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases[J]. Kidney Int, 2021, 100(4S): S1-S276. |
| [25] |
Yazılıtaş F, Kargın Çakıcı E, Kurt Şükür ED, et al. C3 glomerulopathy: experience of a pediatric nephrology center[J]. Acta Clin Belg, 2021, 76(4): 253-257.
doi: 10.1080/17843286.2020.1713450 |
| [26] |
Riedl Khursigara M, Chung E, Tjon J, et al. Utilizing therapeutic drug monitoring to optimize therapy with eculizumab and mycophenolate mofetil in a child with C3 glomerulonephritis[J]. Pediatr Nephrol, 2023, 38(10): 3483-3487.
doi: 10.1007/s00467-023-05927-9 pmid: 36884075 |
| [27] |
Le Quintrec M, Lapeyraque AL, Lionet A, et al. Patterns of clinical response to eculizumab in patients with C3 glomerulopathy[J]. Am J Kidney Dis, 2018, 72(1): 84-92.
doi: S0272-6386(17)31139-3 pmid: 29429752 |
| [28] |
Duineveld C, Steenbergen EJ, Bomback AS, et al. Treatment-resistant nephrotic syndrome in dense deposit disease: complement-mediated glomerular capillary wall injury?[J]. Pediatr Nephrol, 2020, 35(9): 1791-1795.
doi: 10.1007/s00467-020-04600-9 pmid: 32447506 |
| [29] |
Welte T, Arnold F, Westermann L, et al. Eculizumab as a treatment for C3 glomerulopathy: a single-center retrospective study[J]. BMC Nephrol, 2023, 24(1): 8.
doi: 10.1186/s12882-023-03058-9 pmid: 36631797 |
| [1] | NIU Yanhua, DONG Xiaoyan. Clinical characteristics and risk factors for plastic bronchitis in children with macrolide-resistant Mycoplasma pneumoniae pneumonia [J]. Journal of Clinical Pediatrics, 2026, 44(2): 111-117. |
| [2] | LI Yun, GAO Hongbo, DI Yuqing, LI Longmin, LIU Haichun, ZHANG Xiaoyong, SHAO Yujun. Clinical characteristics and prognosis of metastatic pheochromocytoma and paraganglioma in children and adolescents [J]. Journal of Clinical Pediatrics, 2026, 44(2): 118-123. |
| [3] | LI Ming, YUE Tong, WEN Min, WU Feifei, ZHANG Dan, XU Yingjie, KANG Min, ZHU Jia, YAN Yuchun, LAI Jianming, WU Fengqi. Clinical characteristics of Behcet syndrome with cardiovascular aneurysms in children [J]. Journal of Clinical Pediatrics, 2026, 44(2): 146-153. |
| [4] | LIANG Haodong, GUO Shengxuan, HU Siyuan, LU Xuanjun, ZHANG Yunhan, XU Chenxia. Research progress of the symptom scale for acute upper respiratory tract infection in children [J]. Journal of Clinical Pediatrics, 2026, 44(2): 161-166. |
| [5] | Subspecialty Pediatric Consensus Collaborative Group, Subspecialty Group of Infectious Diseases, Society of Pediatric, Chinese Medical Association, Subspecialty Group of Infectious Diseases, Society of Pediatric, Zhejiang Medical Association, National Clinical Research Center for Children and Adolescents' Health and Diseases. Expert consensus on the clinical application and individualized therapy of oxazolidinone antibiotics in children [J]. Journal of Clinical Pediatrics, 2026, 44(1): 1-16. |
| [6] | CHEN Dan, REN Jiaying, SUN Lifang, WEI Zhenghu, SUN Xiaomin. Clinical features and prognosis of severe Mycoplasma pneumoniae pneumonia combined with intracardiac thrombus in children: a single-center retrospective study [J]. Journal of Clinical Pediatrics, 2026, 44(1): 17-24. |
| [7] | LIU Min, WANG Qi, SU Jun, CUI Lidan, SUN Huixia, NING Wenhui. Clinical features, diagnosis, and treatment of Mycoplasma pneumoniae pneumonia secondary to infective endocarditis in children [J]. Journal of Clinical Pediatrics, 2026, 44(1): 25-30. |
| [8] | JIA Ru, MA Yan, YU Qun, LIU Haiping, LIU Ping. An analysis of influenza vaccine coverage and influencing factors among children under 16 years old in two suburban districts of Shanghai [J]. Journal of Clinical Pediatrics, 2026, 44(1): 31-37. |
| [9] | WU Jinjun, XIONG Hao, ZENG Hui, CHEN Zhi, YANG Li, SUN Ming, WANG Zhuo, DU Yu, QI Shanshan, WANG Wei, ZHANG Lannan. Clinical analysis of allogeneic hematopoietic stem cell transplantation on pediatric myeloid sarcoma [J]. Journal of Clinical Pediatrics, 2026, 44(1): 44-50. |
| [10] | LIANG Zhiru, GUO Linmei, WANG Fei, ZHAO Xiaoyun. Lupus enteritis or inflammatory bowel disease: a report of the diagnosis and treatment of systemic lupus erythematosus complicated with gastrointestinal symptoms [J]. Journal of Clinical Pediatrics, 2026, 44(1): 56-63. |
| [11] | ZHOU Yuanke, SUN Lina, FANG Ying. Research progress on the role of zinc in the occurrence, development and treatment of inflammatory bowel disease in children [J]. Journal of Clinical Pediatrics, 2026, 44(1): 64-70. |
| [12] | ZHANG Wenting, FENG Juan. Progress in pediatric Takotsubo syndrome [J]. Journal of Clinical Pediatrics, 2026, 44(1): 79-83. |
| [13] | LIU Yun, PAN Jingjing, SHEN Jinxin, ZOU Yunsu, CHENG Rui, FENG Yun, YANG Yang. Therapeutic efficacy of surfactant supplementation after pulmonary hemorrhage in improving clinical outcomes of preterm infants with gestational age <34 weeks [J]. Journal of Clinical Pediatrics, 2025, 43(9): 652-660. |
| [14] | ZHANG Wei, WANG Yang, DENG Wenhua, WU Yabin. Analysis of clinical manifestations, ciliary structure and genetic characteristics of primary ciliary dyskinesia in 14 children [J]. Journal of Clinical Pediatrics, 2025, 43(9): 680-685. |
| [15] | XIANG Linjuan, CHEN Xuexin, JIA Yanhui, WU Yuhang, CONG Xin, LI Wei, CHEN Yingying, CHEN Sun, HUANG Lisu. Prognostic factors analysis of adenovirus type 3 pneumonia in children [J]. Journal of Clinical Pediatrics, 2025, 43(9): 686-691. |
|
||